
Reactions were mixed some 14 years ago, when we and others first reported that in rodent models of type 1 diabetes (T1D), metabolic derangements resulting from severe insulin deficiency could be fully reversed by infusing the adipocyte (fat cell) hormone leptin directly into the brain. A complete departure from our widely accepted understanding of how glucose homeostasis works, this finding was met with comments like “That makes no sense – how is it even possible???”. The difference between a finding that is hailed as a scientific breakthrough and one that is set aside because it makes no sense often comes down to the context within which it is understood. At the time, there was simply no way to understand how leptin action in the brain could make the most severe, potentially lethal consequences of diabetes simply disappear, without the need for insulin.
In the years since, our understanding of the brain’s role in the manifestations of uncontrolled diabetes has matured considerably. Of particular importance is the interaction between the brain, which drives fuel-mobilizing responses when it perceives that body fuel stores are under threat, and the pancreatic hormone insulin, which limits mobilization of these fuels. This interaction is fundamental to survival under conditions when food is not readily available, but it can go completely haywire if there’s not enough insulin present to limit fuel mobilization. The goal of this article is to provide a modern framework for understanding how the brain, under the influence of leptin, can normalize glycemic control without the need for insulin. The place to begin this scientific journey is with an overview of how the body’s fuel needs are met when food is no longer present in the GI tract.
Meeting the body’s fuel needs when food is not available
Ensuring that the body’s fuel needs are continuously met is a complex challenge. This is particularly true for mammals that consume food in discrete ‘meals’, which means that nutrients are entering the body from the GI tract only part of the time. After ingested nutrients are absorbed, stored fuels must be mobilized to meet the body’s ongoing energy requirements. Indeed, survival depends on the ability to ensure that body fuel requirements are met during periods when food is not available, and this goal is achieved by complex interactions between the brain and peripheral organs, including the pancreas, liver, and fat (adipose) tissue.
The brain has the unique ability to detect when body fuel stores are threatened, and the unique responsibility to ensure that when this happens, fuels are mobilized in amounts that meet ongoing energy needs. The primary fuels involved are glucose and ketones, which are released into the bloodstream from the liver, and free fatty acids (FFA) and glycerol, which are released through the breakdown of stored fat (a.k.a., triglycerides) in a process known as lipolysis. Virtually all cells in the body can survive so long as these fuels are available in amounts sufficient to meet their energy needs.
How does the brain trigger fuel mobilization?
The brain has an impressive array of tools with which to turn on fuel mobilization. Among these are the release of hormones such as growth hormone, cortisol, and epinephrine. Also important are changes in the activity of the autonomic nervous system, particularly the sympathetic nervous system (SNS). Increasing the activity of SNS nerves supplying the liver, for example, promotes the production and release of glucose and ketones, while activation of SNS outflow to adipose tissue stimulates lipolysis, thus releasing FFAs and glycerol into the circulation. These fuel-mobilizing responses are interconnected in the sense that in the liver, FFAs are the primary building blocks for ketone synthesis, and glycerol can be used to make glucose.
An equally important but opposite role is played by the hormone insulin, which is secreted from so- called ‘beta cells’ located in the pancreas. Insulin secretion is stimulated by glucose and other nutrients (in a manner that can be dialed up or down by many different inputs, including autonomic input from the brain). In turn, insulin acts on the liver to block the production of glucose and ketones, and it acts in adipose tissue to block lipolysis. It therefore follows that if insulin levels are too high, fuel mobilization is inhibited, whereas if insulin is unable to be secreted because of damage to pancreatic beta cells, the normal brake on fuel mobilization is lifted and the floodgates open up, so to speak. As we will see, this sequence of events is central to the pathogenesis of the most serious acute complication of uncontrolled T1D, known as diabetic ketoacidosis (DKA).
How does the brain know whether body fuel stores are depleted?
Information regarding the status of body fuel stores is conveyed to the brain by hormones and nutrients in the circulation and by neural signals coming from the vasculature and the GI tract. Of particular importance to the brain are the circulating levels of insulin as well as the adipocyte hormone leptin. Leptin is secreted by adipocytes when fat is being deposited, but this secretion comes to a rapid halt when stored fat is being broken down by lipolysis. Thinking back to the fact that fasting lowers the insulin level, and a low insulin level releases the brake on lipolysis, it follows that when circulating insulin levels are low, so too are leptin levels. Thus, fasting is characterized by low levels of both hormones, and the brain interprets this combination as evidence that body fuel stores are under threat.
The brain can also detect the circulating level of glucose, as will be discussed later in this article. It is rare for the blood glucose level to drop below its biologically defended level (known as hypoglycemia), but when this happens, the brain rapidly engages responses that return the glucose level to the normal range. The reason that hypoglycemia is so rare in normal animals is that the brain is extremely good at mobilizing glucose before the blood level gets too low. The primary context in which hypoglycemia occurs is when insulin is given to patients with diabetes at a dose that drops the blood glucose level out of the normal range.
Another truism is that both leptin and insulin circulate at levels proportional to body fat content. In response to fasting, however, the level of both hormones quickly falls, constituting a powerful signal that body fuel stores are under threat. Yet even in the face of high circulating glucose and insulin levels, the brain interprets a low leptin level as evidence of depleted fuel stores. Stated differently, brain detection of a low leptin level trumps pretty much everything else when it comes to assessing the status of stored fuel.
With this background, we can now consider homeostatic control of fuel metabolism through a somewhat wider lens, before discussing how the process is disrupted in patients with uncontrolled T1D.
Integrated central and peripheral control of fuel metabolism
Most physiological systems rely upon continuous, dynamic, two-way communication between the brain and peripheral tissues, and the control of fuel metabolism is no exception. Before, during, and in between meals, signals generated by the GI tract, pancreas, liver, and fat (adipose) tissue are transmitted to the brain. The information conveyed by these signals is relevant to 1) the status of stored fuel, 2) the amount of circulating fuel that is available to meet ongoing tissue requirements, and 3) whether nutrients are – or soon will be – entering the body.
The latter information is conveyed via so-called ‘gut-brain’ signals. These signals come in many forms, ranging from autonomic and sensory nerves supplying the GI tract to the release of ‘gut peptides’ such as GLP1 and CCK. Rising levels of various nutrients in the bloodstream can also be detected by the brain. In response to these various signals, the brain modulates a wide array of bodily functions, including stimulation of various digestive processes, secretion of metabolic hormones (including insulin), and helping to coordinate whether the liver will consume or produce glucose. This system for two-way communication is collectively referred to as “the gut-brain axis.”
In addition to low levels of leptin and insulin, the absence of nutrient-induced gut-brain signals also communicates the fasted state to the brain. Combined with the effect of fasting to lower the blood glucose, the fasting brain perceives that 1) circulating fuel levels are on the decline, 2) stored fuel is also under threat, and 3) food is no longer present in the GI tract. Time to sound the alarm!
In turn, the brain mounts responses to ensure that body energy needs are met while seeking out food in the environment and minimizing any unnecessary expenditure of energy. Here is a more specific list:
- Promote production of glucose and ketones by the liver.
- Stimulate lipolysis, such that FFAs and glycerol mobilized from fat stores become an additional fuel source.
- Stimulate all aspects of feeding – the motivation to find and consume food, while increasing its rewarding and decreasing its satiating properties.
- Increase metabolic efficiency by reducing heat production.
- Suppress reproductive function. (At issue here is that attempting to reproduce while struggling to meet your own energy requirements is maladaptive.)
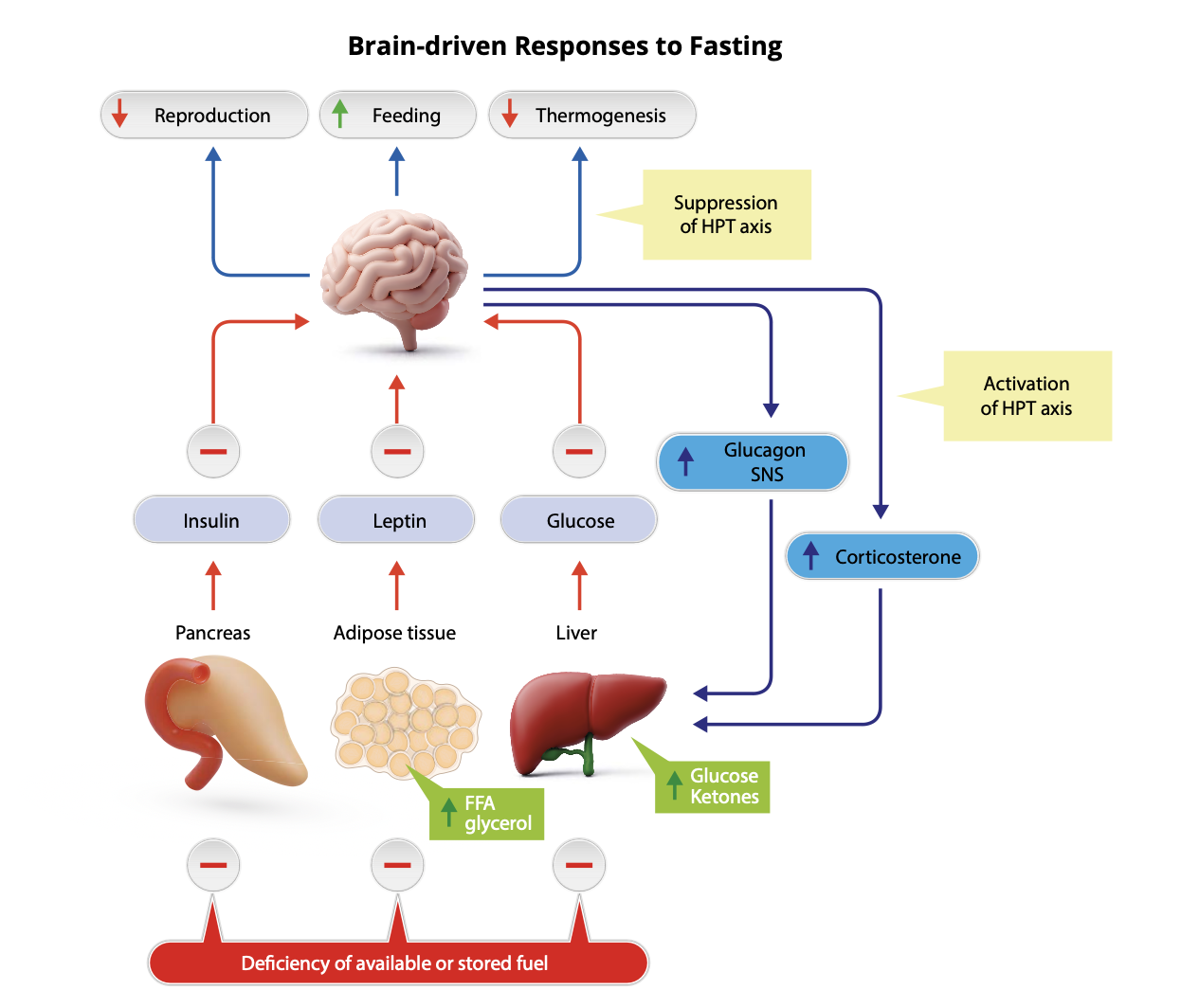
How does the brain coordinate these multi-pronged adaptive responses? Here is another list:
- Activation of the hypothalamic-pituitary adrenal (HPA) axis to increase secretion of the stress hormone cortisol (in rodents, its corticosterone).
- Increase SNS outflow to adipose tissue, liver and pancreas, thereby stimulating lipolysis as well as liver production of glucose and ketones, while also suppressing insulin secretion.
- Suppress the secretion of reproductive hormones.
- Suppress the secretion of thyroid hormone.
- Activate neurocircuits that drive feeding behavior
At risk of redundancy, it should be stressed that interactions between brain and periphery are required to effectively mount these responses. For example, brain-driven autonomic and neuroendocrine responses that mobilize fuel are antagonized by the action of insulin, so fuels cannot be effectively mobilized unless the circulating insulin level is low.
Stated differently, effective fuel mobilization requires that the brain’s perception of fuel deficiency be combined with a low circulating insulin level. At the same time, the nutrient-responsive nature of insulin secretion ensures that fuels are not mobilized in an unrestrained manner. In normal animals, fasting is characterized by a steady state wherein the rate at which fuels enter the circulation is matched by the rate at which they are utilized, such that blood levels do not exceed normal limits.
It is also worth reiterating that among the various inputs that the brain processes as it mounts responses to fasting, a low leptin level has an outsized impact. Even in the face of increased fat mass and elevated circulating levels of insulin and glucose, the brain interprets leptin deficiency as evidence of deficient fuel stores, and most of the adaptive responses listed above will be engaged. Consequently, genetic leptin deficiency in both humans and animals can result in both obesity and diabetes because the brain falsely perceives the unrelenting need to consume and mobilize fuels.
Control of insulin secretion
While glucose is the primary driver of insulin secretion by pancreatic beta cells, the insulin response to glucose is highly regulated, with autonomic input from the brain playing an important role. For example, the stimulatory effect of glucose on insulin secretion can be completely blocked if SNS outflow to the pancreas is increased. Conversely, consuming a meal increases parasympathetic input to the pancreas (via the vagus nerve), and this augments insulin release.
How does insulin lower the BG level? In two primary ways. First, by promoting the movement of glucose from the bloodstream and into peripheral tissues and second, by suppressing its production and release into the circulation (primarily by the liver, but also by the kidney). Each of these effects involve binding of insulin to the insulin receptor, and most of the downstream intracellular signaling events have been worked out.
The comparative importance of the effects of insulin to promote glucose disposal vs. glucose production depends on the nutritional state. During and following a meal, insulin secretion increases, and insulin-mediated glucose disposal becomes a predominant determinant of glucose homeostasis. During fasting, however, insulin levels are too low to effectively promote glucose uptake into tissues. Instead, its primary role is to limit the rate at which nutrients – glucose, ketones, FFAs, and glycerol – are released into the circulation. This action of insulin provides an important counterweight to brain-driven mobilization of these substrates. Ultimately, the circulating level of these various nutrients during fasting is determined by the balance between the effect of the brain to promote and the effect of insulin to limit their mobilization.
How does uncontrolled T1D impact this control system?
In T1D, pancreatic beta cells are destroyed by the immune system, which mistakenly perceives them to be foreign invaders. The disease begins more commonly in children or adolescents than in adults, but it can develop at any age. The resulting severe insulin deficiency sets in motion a series of events with potentially catastrophic consequences. On the one hand, insulin deficiency reduces the body’s ability to move glucose into tissues from the bloodstream. On the other hand, it permits excessive mobilization of glucose, ketones, FFAs, and glycerol, for reasons described earlier.
These problems are compounded by the fact that leptin secretion (by fat cells) is dependent on insulin; leptin levels also drop precipitously, a powerful signal to the brain that fuel stores are depleted. So long as leptin levels remain low, the brain will perceive fuel deficiency regardless of how much fuel actually exists, and as long as insulin levels are low, fuel mobilization cannot be slowed. Furthermore, leptin deficiency is a powerful stimulus to eat, and excessive food intake is a cardinal feature of uncontrolled diabetes (referred to as ‘diabetic hyperphagia’). Consequently, fuel-mobilizing machinery is activated, robustly and relentlessly, with no way to stop it. It’s the equivalent of stepping on the fuel mobilization accelerator pedal while taking your foot off the brake, with everything compounded to excessive caloric intake.
As the circulation becomes flooded with glucose, ketones, FFA, and glycerol, new problems set in. For one, elevated levels of these nutrients begin to have a dehydrating effect, which in turn shrinks the circulating blood volume. This begins to limit the ability to provide the blood supply that kidneys and other organs need to function properly. Making matters considerably worse, ketones are released from the liver in the form of an acid (ketoacids), so this cascade progresses, the blood becomes acidic. Over time, this becomes a serious threat to the carefully regulated acid-base balance upon which normal cellular function depends. This progression of dehydration and acidosis poses a serious threat to survival.
Responding to this threat, the brain does what it does under most stressful situations: it activates the sympathoadrenal system, releasing epinephrine (adrenalin) into the circulation. Unfortunately, epinephrine also drives fuel mobilization, effectively throwing fuel on the fire. The result is a vicious cycle wherein dehydration and acidosis trigger stress responses that exacerbate the underlying fuel mobilization crisis, causing more and more dehydration and acidosis. This is the condition known as diabetic ketoacidosis (DKA), and if untreated, death ensues.
A century ago, the discovery of insulin was heralded as a miracle and rewarded with a Nobel Prize because its administration could halt the unrestrained metabolic derangements that drive DKA. Indeed, it has been thought of ever since as the only way to achieve this outcome.
But there’s another way to treat DKA, and that is the main point of this article. Based on the above considerations, logic dictates that if the brain does not perceive that fuel stores are depleted, or if the brain is rendered unable to turn on the fuel-mobilizing machinery, this should prevent the runaway train of severe metabolic impairment that results in DKA.