
Researchers are investigating the continued ineffectiveness of amyloid-targeting therapies which points to either flaws in the amyloid cascade hypothesis, or its application in drug development – but what can be done to help? And what can ARC do?
Alzheimer’s disease is a progressive neurological disorder characterized by the gradual deterioration of memory, thinking, behavior, and the ability to perform everyday activities. It is the leading cause of dementia, primarily affecting older adults. Symptoms typically begin with short-term memory loss and progress to disorientation, language difficulties, impaired judgment, and personality changes. Concurrently, physical changes occur in the brain. Early on, synaptic connections become dysfunctional and are lost, contributing to cognitive decline. In advanced stages, neurodegeneration leads to brain atrophy, particularly in areas crucial for memory and executive function, such as the hippocampus and cortex.
Alzheimer’s disease (AD) currently affects over 40 million people worldwide. As life expectancy increases, AD prevalence is projected to rise sharply, reaching epidemic levels and posing a significant public health and economic burden. Dementia cases are projected to nearly triple by 2050 – reaching 140-150 million, with AD comprising the majority. No therapy currently prevents, stabilizes, or reverses its progression. Despite extensive research – encompassing over 2 million scientific papers and more than 3,000 clinical trials that have tested 138 different drugs – none have successfully halted or reversed the disease.
What accounts for this failure? Many researchers argue that the continued ineffectiveness of amyloid-targeting therapies points to flaws in the amyloid cascade hypothesis, or its application in drug development.
The Amyloid Cascade Hypothesis
The story of the amyloid hypothesis starts in 1906, when a German psychiatrist by the name of Alois Alzheimer identified the first case of the debilitating disease named after him. Dr. Alzheimer described neurofibrillary tangles and amyloid plaques in his patient’s brain as unique hallmarks of her dementia. This initial assessment resulted in the amyloid hypothesis for AD, which posits that the accumulation of amyloid-β (Aβ) peptides – especially Aβ42 – leads to plaque formation, triggering downstream events, including tau hyperphosphorylation, neuroinflammation, synaptic loss, and neuronal death.
The hallmark physical changes associated with Alzheimer’s include the accumulation of amyloid plaques and neurofibrillary tangles in the brain. These plaques, composed of aggregated amyloid- beta proteins, disrupt communication between neurons. Meanwhile, neurofibrillary tangles, formed from tau protein, impair the transport of essential nutrients within neurons, ultimately leading to cell death. These pathological features create a cascade of events that exacerbate neuroinflammation and oxidative stress, further compounding the damage to brain tissue. This theory has driven most AD drug trials over the past 20+ years.
The amyloid cascade hypothesis became the dominant – almost monopolistic – paradigm in AD drug discovery due to a convergence of compelling early evidence, strong institutional momentum, and the lack of competing frameworks with similar explanatory power in the 1990s and early 2000s. The amyloid cascade hypothesis was first formally articulated in 1992 by John Hardy and Gerald Higgins. It was primarily grounded in genetic findings. Mutations in the Amyloid Precursor Protein (APP) in familial AD directly increased amyloid-β42 production. Presenilin-1 and Presenilin-2 mutations (components of γ-secretase) were shown to shift cleavage of APP toward more aggregation-prone Aβ42. These findings suggested a causal role for amyloid accumulation in early-onset familial AD, and this was extrapolated to sporadic, late-onset AD, despite fundamental differences in pathogenesis.
Histopathological hallmarks also supported the model. Postmortem brains of AD patients showed abundant extracellular amyloid plaques and intracellular tau tangles. The progression of amyloid pathology seemed to match clinical decline, at least in early interpretations. Mouse models overexpressing mutant APP or presenilin recapitulated amyloid pathology. These models became standard preclinical tools, reinforcing the centrality of Aβ. However, they rarely showed tau tangles, neurodegeneration, or cognitive decline unless manipulated further – yet this limitation was often overlooked.
But is it a red herring?
However, there are also critical problems associated with the amyloid hypothesis. Treatments that remove amyloid do not result in cognitive recovery. Drugs like aducanumab (currently discontinued) and lecanemab do clear amyloid plaques, as shown by PET imaging. However, their cognitive benefit is minimal or absent. This disconnect raises questions about whether amyloid is a cause, a consequence, or even a protective response. Amyloid plaques are also found in cognitively normal individuals. Up to 30% of elderly individuals have significant amyloid deposition but no cognitive impairment. This suggests that plaque burden alone may not be sufficient to cause dementia. Symptoms correlate more closely with tau pathology and synaptic loss. Tau tangles and synaptic degeneration show a stronger correlation with cognitive decline than amyloid burden. This supports the idea that amyloid may trigger disease, but downstream events drive symptoms.
It therefore appears that the failure of several thousand clinical trials to identify a therapy that halts or reverses AD has been caused by the scientific community’s commitment to devote all their time and money to the amyloid cascade hypothesis, which may be a red herring. Although there is a strong correlation between amyloid dysfunction and cognitive decline in AD, it appears that there is no causative relationship. Unfortunately, the strict adherence to the amyloid cascade hypothesis has hindered the exploration of alternative theories. So, how are we to proceed from here?
Alzheimer’s disease is a system-level disorder
Alzheimer’s disease (AD) is increasingly recognized as a system-level disorder, rather than a condition caused by a single pathological event like amyloid-β accumulation. It involves multiple brain regions, many molecular cascades, and divergent cell types, all interacting over time in a complex, dynamic network of dysfunction. AD affects numerous interconnected molecular and cellular pathways:
- Proteinopathy: protein folding and aggregation problems, including amyloid-β (extracellular plaques), tau (intracellular tangles), and TDP-43.
- Neuroinflammation: chronic activation of microglia and astrocytes, and cytokine dysregulation (e.g., IL-1β, TNF-α).
- Synaptic dysfunction: loss of synaptic plasticity, glutamate excitotoxicity, Arc dysregulation.
- Mitochondrial failure: impaired energy metabolism, oxidative stress, defective mitophagy.
- Lipid and membrane metabolism: ApoE4- driven lipid dysregulation, aberrant cholesterol trafficking.
- Vascular pathology: blood-brain barrier breakdown, reduced perfusion, microinfarcts.
- Calcium signaling: ER stress, disrupted calcium homeostasis, neuron-glia communication failure.
- Epigenetic alterations: chromatin remodeling, histone acetylation/methylation, altered gene expression.
In summary, AD is a complex system-level disorder involving the progressive breakdown of neural networks, synaptic integrity, and cellular homeostasis across multiple brain regions. Its pathogenesis arises from the interaction of diverse molecular pathways rather than from a single pathogenic trigger. Because AD is a multi-hit, system-level disease, single-target therapies (like anti-amyloid drugs) are likely to be insufficient. Future therapeutic strategies will need to target multiple nodes in the disease network (e.g., inflammation, synaptic repair, metabolism). How do we develop a therapeutic approach that targets the gene networks and signaling pathways that cause the pathogenesis of AD? A recent finding from our laboratory supports this direction. It centers on the memory gene Arc, which is a master controller of both synaptic transmission and neuronal gene expression.
Arc: A therapeutic hub for Alzheimer’s disease
Arc (Activity-Regulated, Cytoskeleton-associated protein) is a key regulator of synaptic function and neuronal plasticity. Removing the Arc gene in mice results in a specific loss of long-term memory, underscoring its crucial role in memory consolidation. Arc is enriched in excitatory synapses where it controls endocytosis of the AMPA subtype of glutamate receptors, allowing it to regulate synaptic plasticity. We found that Arc also localizes to the neuronal nucleus, where it interacts with highly dynamic chromatin. Arc’s nuclear distribution pattern and dynamic behavior suggest it could play a role in the epigenetic control of gene expression.
Arc has already been linked to AD: it is necessary for the formation of amyloid plaques, Arc protein levels are abnormally regulated in the hippocampus of AD patients, and a polymorphism in the Arc gene reduces the risk of developing AD. The connection to AD was considerably strengthened with our recent finding that Arc controls the expression of most Arc susceptibility genes and more than 100 genes implicated in the pathophysiology of AD. Arc regulates transcription of genes involved in synaptic plasticity, memory formation, intrinsic excitability, receptor signaling pathways, and neuromodulation. Targeting Arc opens a new frontier in “multi-target” AD therapy, designed to intervene in multiple aspects of the disease simultaneously, thereby serving as a therapeutic hub.
How can we manipulate Arc’s function? There are two general approaches: (i) regulating the transcription of Arc itself, and (ii) targeting the gene networks that Arc regulates.
Controlling Arc transcription
We had earlier discovered that neuronal activity- dependent Arc transcription is epigenetically controlled by a chromatin-modifying complex containing the histone-modifying enzymes Tip60 and PHF8. Tip60 is a histone deacetylase. Studies in a fruit fly AD model suggest that modulating Tip60 activity can counteract neurodegenerative processes associated with AD. PHF8 is a histone demethylase also implicated in AD: decreased PHF8 levels lead to down-regulation of autophagy, which impairs the cell’s ability to clear amyloid-beta (Aβ) peptides, leading to their accumulation – a hallmark of AD. The finding that PHF8 and Tip60 together control the transcription of Arc highlights the potential of targeting this epigenetic pathway for developing AD therapeutics. We have therefore developed drugs targeting these two enzymes. High-resolution X-ray structures are available for both Tip60 and PHF8, in complex with their respective substrates. We have used computer docking to identify novel compounds that bind to the substrate binding sites, thereby interfering with their enzymatic activity. The efficacy of these drugs to inhibit neuronal activity induced Arc expression was tested using neuronal networks of cultured rat hippocampal neurons. The figure shows 15 commercially available compounds that significantly inhibited Arc induction. Ordering information is found in the Zinc15 database using the zinc IDs shown in the figure above.
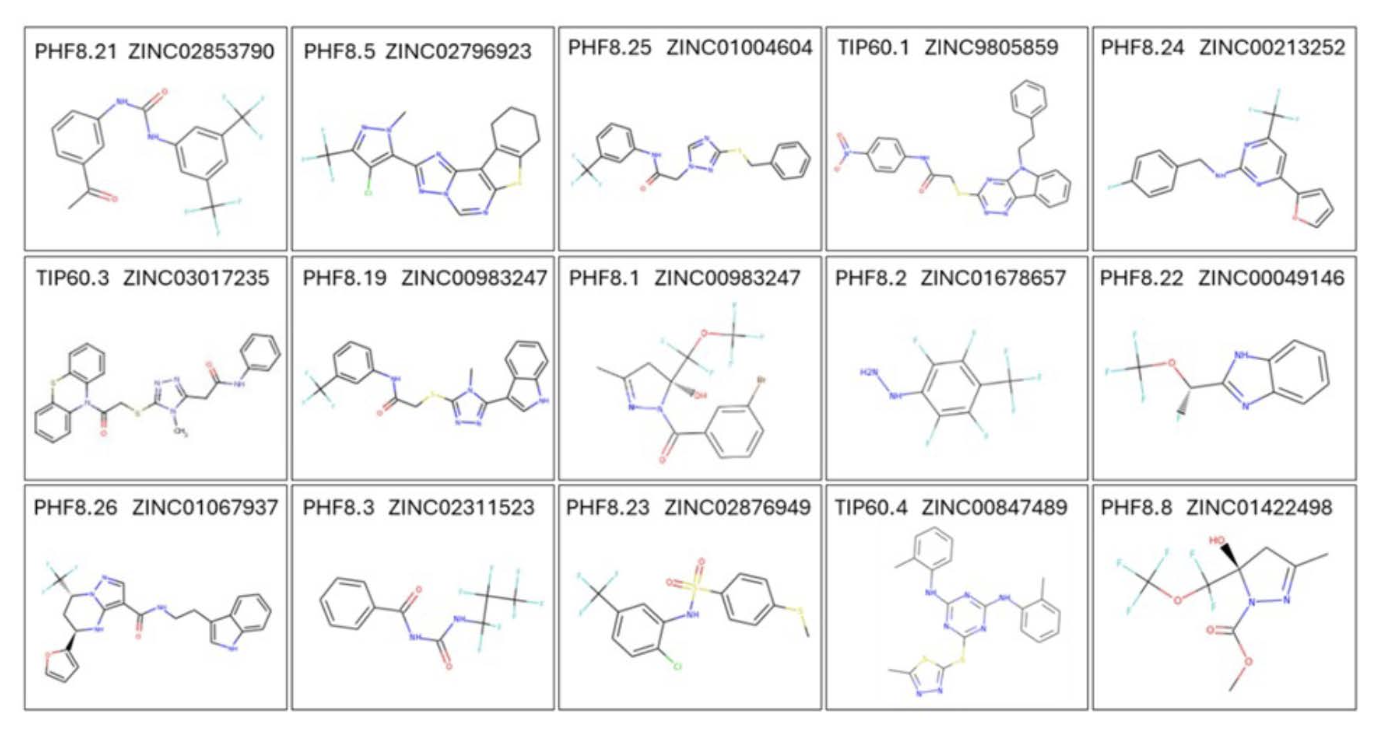
These compounds require further evaluation for their ability to cross the blood-brain barrier. The main question that remains to be answered is: Does the inhibition of activity-dependent Arc expression halt AD progression? This can be addressed with experiments in a triple transgenic AD mouse model. At this point, it isn’t clear whether manipulation of Arc expression can affect AD outcome. It is possible that enhancing Arc’s function is therapeutic, while inhibiting Arc is deleterious or ineffective. Therefore, a second approach is needed to test this possibility. Let’s look at that next.
Targeting the gene networks controlled by Arc
Arc regulates transcription of more than 100 genes implicated in the pathophysiology of AD. One set of genes is transcriptionally activated upon arc induction, while functional Arc inhibits transcription of the other set. Since it is unclear at this point whether Arc has a positive or negative effect on AD progression, it would be beneficial to have a strategy that can be developed into a therapy for each of these possible scenarios.
This can be accomplished by targeting one of the two gene networks that are positively and negatively affected by Arc. This strategy is made possible by drug promiscuity: the ability of drugs to bind to more than one protein/receptor with high affinity. We have recently performed a pilot study on drug promiscuity, which demonstrated the feasibility of this approach for drug discovery. Using molecular docking, we will explore this avenue by estimating the binding affinities of all FDA-approved drugs for the proteins encoded by the AD gene networks controlled by Arc. The goal is to identify drugs that selectively bind to multiple proteins in each network with high affinity. A study is planned to expand the database to include ten million commercially available drug-like compounds. The drugs identified in these in silico screens then need to be tested for their efficacy in curbing disease progression in an AD mouse model.
