This article discusses research by Dr. Lee J. Martin and his team on HIE, a leading cause of neonatal mortality. They use human induced pluripotent stem cells (iPSCs) and neural stem cells (NSCs) and emphasize the vulnerability of oligodendrocytes, sharing how these cells can accumulate toxic misfolded proteins, potentially causing severe neural damage and long-term cognitive disabilities in affected infants
Hypoxic-ischemic encephalopathy (HIE) is a significant cause of mortality and morbidity in infants. Birth asphyxia causes nearly one million neonatal deaths worldwide annually. Therapeutic opportunities are very limited for HIE babies. Mild hypothermia (HT) reduces the risk of death or disability. It is the standard of clinical care for term pregnancy HIE in Western countries, but ~50% of the infants treated with HT die or suffer persistent and potentially lifelong neurologic and cognitive disabilities. Specific neural systems and populations of cells in the brain are selectively vulnerable in HIE.
Understanding Oligodendrocytes
Dr. Lee J. Martin and his group of researchers at Johns Hopkins University School of Medicine have studied neonatal HIE for decades. One of these populations of vulnerable cells is called oligodendrocytes (Fig. 1A). They are enriched in white matter but are also present in gray matter. Oligodendrocytes have many functions in the brain. One main function is axon myelination (Fig. 1A). Axons connect neurons together in the central nervous system (CNS) and organs and skeletal muscles in the peripheral nervous system.
Oligodendrocytes make myelin for axons in the CNS (Fig. 1A), allowing them to modify their electrical properties and sustain growth factor support. Axons without their proper myelin do not convey their electrical signals properly and can degenerate. The signals propagated by axons are called action potentials. Oligodendrocyte and white matter damage in the brain can be detected by brain MRI in some experimental animal models of neonatal brain injury. They could be in part responsible for some of the neural network damage and lifelong neurologic consequences seen in infant survivors of HIE.
Toxic conformer proteins (TCPs)
A recent study from Martin’s group (Martin et al., 2025) discovered in postmortem autopsy brain of infants that died from HIE, and in newborn pig models of HIE, that oligodendrocytes can accumulate abnormal proteins that are misfolded, aggregated, or modified deleteriously at tyrosine amino acid residues (3-nitrotyrosine) by a damaging free radical called peroxynitrite (ONOO-), formed by the combination of superoxide (O2.) and nitric oxide (NO) (Fig. 1G). Several of these abnormal proteins are believed to be toxic and are thus called toxic conformer proteins (TCPs). Some of these TCPs are infamous for causing Parkinson’s disease, Lewy body dementia, amyotrophic lateral sclerosis (ALS), and even Creutzfeldt-Jakob disease because they can kill neural cells, including oligodendrocytes. Our discovery in the neonatal HIE brain was surprising and novel, demonstrating that TCPs could form rapidly, independent of chronological aging and without known underlying gene mutations.
Challenges in studying the formation of TCPs
This new observation is challenging to pursue experimentally. While we found very important evidence in human brain clinical specimens, it is difficult to address questions about how TCPs form in these autopsy specimens because they yield static information about a pathological process with an uncertain onset. Our piglet models of neonatal HIE (Martin et al., 2025) will be incredibly valuable for follow-up experiments. However, these experiments are very costly due to resource and skilled personnel requirements, as well as ethical and societal concerns about using neonatal piglets, which the US Department of Agriculture and Institutional Animal Use and Care Committee closely regulate. Moreover, animal models might not accurately reflect human injury or disease states from a cellular and molecular standpoint (Martin & Zhiping, 2004), which makes disease target discovery and therapeutic development challenging. Studies have shown that DNA damage response (DDR), DNA repair, and cell death mechanisms differ in human neurons compared to mouse neurons (Martin & Chang, 2018). The uniquely human neural cell injury response, DDR, proteasome regulation, and cell death mechanisms represent a paradigm shift for experimental neuropathology and the modeling of human CNS injury and disease. Our solution was to use human induced pluripotent stem cells (iPSCs) and neural progenitor cells (NPCs).
iPSC- and NPC-derived oligodendrocytes to test TCP formation
We used human iPSC- or NPC-derived oligodendrocytes (Fig. 1) and neurons (Fig. 2) to directly test the hypothesis that injury to cells of the human neonatal, immature, or developing brain can cause the formation of TCPs. iPSCs are a type of cell that originates from an adult cell, such as a skin fibroblast or a peripheral blood mononuclear cell, and is genetically reprogrammed in a culture dish using specific genes (Oct4, Sox2, Nanog, and Lin28) introduced by genome-integrating or nonintegrating methods or by direct protein transfer. A defining feature is that iPSCs can generate the three primary germ layers (ectoderm, mesoderm, and endoderm), as if they were embryonic stem cells. However, they are not derived from human embryos. NPCs can be derived from embryonic neural stem cells.
Human oligodendrocytes derived from iPSCs are fascinating to observe growing in living cell cultures at immature (Fig. 1B) and mature (Fig. 1C) stages of differentiation. Their identity as mature oligodendrocytes can be verified by markers such as 2,3-cyclic nucleotide 3-phosphodiesterase (CNPase); this enzyme is abundant in myelin-producing cells (Fig. 1D). When human oligodendrocytes are treated with a chemical, quinolinic acid (QA), that simulates a pathological mechanism of neonatal HIE, called glutamate receptor excitotoxicity, they form pathological accumulations of degenerative vacuoles (Fig. 1F, arrows), that are not present in similar cells treated with control solution (Fig. 1E). They form a TCP called nitrated α-synuclein (Fig. 1H) that is present at low levels in control treated oligodendrocytes. Nitrated-synuclein is an abnormal synaptic protein that is known to cause some forms of neurodegenerative disease.
Human neuron degeneration
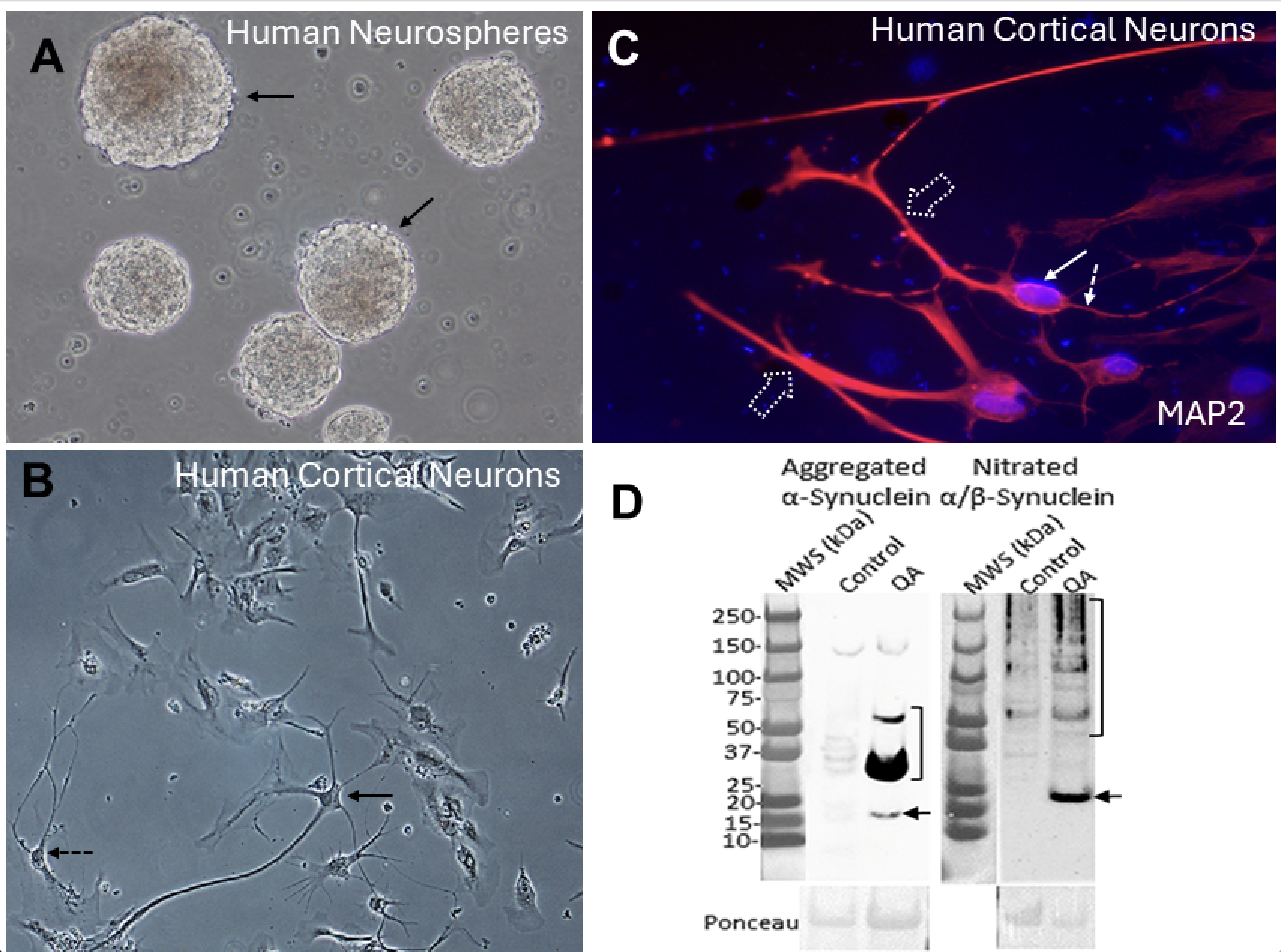
Human neurons generated from iPSCs or NPCs are equally mesmerizing when observed growing in living cell cultures at an immature developmental stage. They form 3-dimensional structures called neurospheres (Fig. 2A). As neurons mature, the neurospheres can disperse and form cerebral cortical-like neurons that differentiate with elaborate cell bodies and delicate processes appearing as large pyramidal-like neurons (Fig. 2B, solid arrow) or interneurons such as arcade or chandelier cells (Fig. 2B, hatched arrow). Their identity as neurons can be verified by a marker such as microtubule-associated protein-2 (MAP2). This cytoskeletal protein is abundant in neurons and can be seen in their cell body (Fig. 2C, solid arrow), dendrites (Fig. 2C, broad dashed arrow), and axon (Fig. 2C, dashed thin arrow). When human neurons are treated with QA, they exhibit a robust degenerative response, characterized by the formation of very high levels of a potent TCP, aggregated α-synuclein (Fig. 2D left), and nitrated αSyn (Fig. 2D right). Control-treated neurons have very low amounts.
αSyn becomes pathological when it is nitrated or aggregated; moreover, some abnormal forms of α-synuclein can cause prion-like spreading and disease in oligodendrocytes and neurons. We found that human oligodendrocytes and neurons rapidly form prion-like forms of α-synuclein in response to excitotoxicity. Clinical and animal studies of neonatal HIE show that there is preferentially protracted brain network-wide degeneration. Neurons, synapses, and oligodendrocytes that accumulate these aberrant proteins could be responsible for the connectome spreading and persistent damage in the neonatal brain with lifelong consequences.
References
- Martin LJ, et al. Hypothermia Shifts Neurodegeneration Phenotype in Neonatal Human Hypoxic-Ischemic Encephalopathy But Not in Related Piglet Models: Possible Relationship to Toxic Conformer and Intrinsically Disordered Prion-like Protein Accumulation. Cells. 2025 Apr 12;14(8):586. doi: https://doi.org/10.3390/cells14080586. PMID: 40277911; PMCID: PMC12025496.
- Martin LJ, Chang Q. DNA Damage Response and Repair, DNA Methylation, and Cell Death in Human Neurons and Experimental Animal Neurons Are Different. J Neuropathol Exp Neurol. 2018 Jul 1;77(7):636-655. doi: https://doi.org/10.1093/jnen/nly040. PMID: 29788379; PMCID: PMC6005106.
- Martin LJ, Zhiping Liu, Opportunities for Neuroprotection in ALS Using Cell Death Mechanism Rationales, Drug Discovery Today: Disease Models, Vol 1, Issue 2, 2004, https://doi.org/10.1016/j.ddmod.2004.09.004.