David Ussery, a Professor in the Department of BioMedical Informatics at UAMS, and his student, Aakash Bhattacharyya, discuss using Machine Learning methods to predict the pathogenicity of a bacterial infection based on genome sequencing
The bacterial genus Salmonella is a common source of foodborne outbreaks, infecting more than one million people in the United States every year. Most of the time, the illness is brief, and recovery occurs within a few days. However, Salmonella infection result in more than 25,000 hospitalizations, and approximately 400 deaths occur each year in the US. There is a need to rapidly determine if a Salmonella infection is likely to be serious, to quickly direct the appropriate medical treatments.
Salmonella was first described by Lignieres in 1901 as ‘le microbe du hog-cholera de Salmon’ (1,2), named after the American veterinary surgeon, Daniel Elmer Salmon. There are more than 2500 different types (‘serotypes’) of Salmonella. Historically, each serotype was named as a species (1) but was then reduced to one species (Salmonella enterica) in 1987 (2) and last revised in 2005 (3) with the addition of another species (S. bongori).
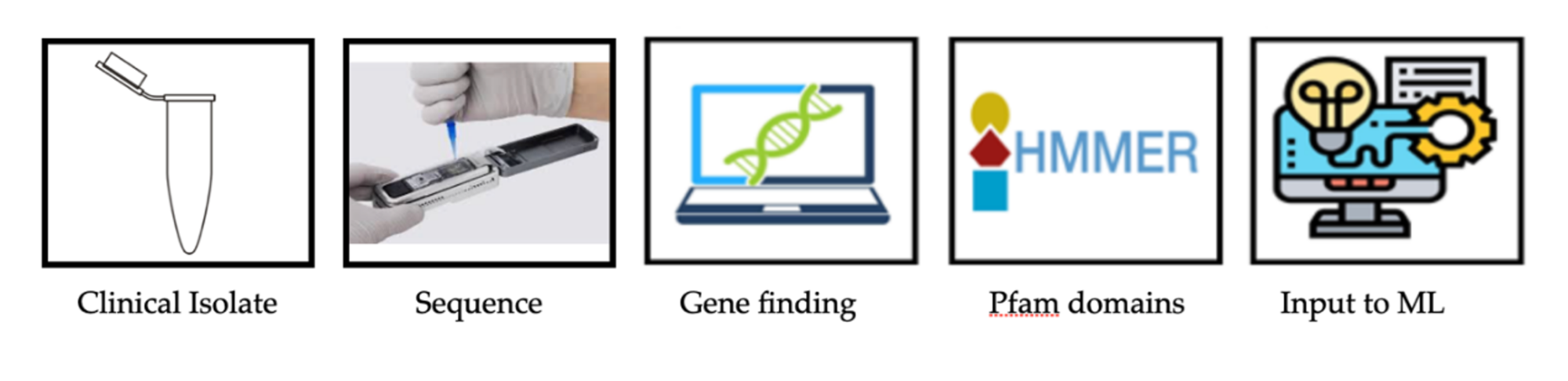
It is now possible, using high-throughput computational methods, to predict the severity of a Salmonella infection, based on the genome sequence of a clinical isolate. In principle, we can transition from a sample to a genome sequence and predict the severity level within a few hours, as shown in the figure above.
AI and disruptive changes in sequencing technology
The first single-molecule or ‘third- generation’ sequencing machine was introduced in 2014 and has dramatically reduced the cost and time required to obtain bacterial genome sequences; this technology enables much longer reads. For bacterial genomes, we routinely obtain approximately 20,000 nucleotides (nt) per read, corresponding to about 20 genes. With careful sample preparation, we can also obtain reads of approximately a million nt from human chromosomal DNA. Oxford Nanopore flow cells sequence single molecules by measuring changes in current as a single strand of DNA passes through a tiny pore of about 1nm (10 atoms) in width – too narrow for double-stranded DNA to fit! This change in current is translated to a sequence, with various Machine Learning methods, such as an artificial neural network trained on known sequences, (4,5) including modified bases, (6) such as 5mC. This disruptive technology allows for rapid and inexpensive sequencing, with the newer version of flow cells (R10.4.1) capable of reading DNA fragments at >99% single read accuracy; (7) the long reads can be used to completely sequence and assemble bacterial genomes and plasmids in a few hours, improving the quality (and quantity) of sequenced genomes. As mentioned in a previous profile article, (8) approximately 1.2 million Salmonella genome sequences were available for comparison as of December 2024. Over the past five months, an additional 200,000 Salmonella genomes have become available (as of May 2025), and the number continues to grow rapidly.
Computational analysis of proteomes
This brings us to the problem of how to quickly analyze literally millions of genome sequences, especially with the aim towards helping medical doctors decide whether a particular strain of Salmonella isolated from a patient is likely to cause severe and possibly fatal disease. Genome sequences can be stored as strings of four letters (GATC) representing the four bases in DNA, but digital computers use numbers, and comparing just letters is obviously not enough. In living Salmonella cells, the genome gets transcribed into RNAs, most of which encode proteins. The ‘information’ is in the protein sequence, but again, this can be thought of as just a string of 20 letters, one representing each amino acid. How does one compute with a string of letters? The basis of this dates back to the 1960s, with the creation of the Atlas of Protein Sequence and Structure, (8,9) which eventually became part of the UniProt database. (10)
Profile HMMs to abstract proteomes
We have described previously how one can use profile HMMs to find functional domains, such as Pfam domains, (11) within all the proteins in a genome, and then use those to quickly pull out a set of specific proteins (such as sigma factors) from thousands of genomes, in just a few seconds. These Pfam domains can be used to search for enrichment in genomes known to cause pathogenicity, and then this information can serve as input for Machine Learning methods.
Ultimately, we identified a set of Pfam domains that could be utilized as biomarkers for accurately predicting the severity of cases in 93% of our test set. The methods described here are just one example out of many – at the time of writing (May 2025), there are more than 200 articles in PubMed when searching for ‘Salmonella Machine Learning.’ The number of publications will likely rapidly grow, as labs around the world continue to apply Machine Learning methods to study Salmonella genomes and pathogenesis.
The project described here has been accepted for publication – new reference: Bhattacharyya A, Panday S, Ussery D. Rapid assessment of clinical severity for salmonellosis cases via protein family domain analysis and machine learning. Academia Molecular Biology and Genomics 2025;Volume. https://doi.org/10.20935/xxx
References
- John-Brooks, E. St., (1934). “The Genus Salmonella Lignieres, 1900”, Journal of Hygiene (London). 34(3):333-350. doi:
- Le Minor, L., & Popoff, M. Y., (1987). “Designation of Salmonella entérica sp. no. nom. rev., as the type and only species of the genus Salmonella”, International Journal of Systematic Bacteriology, 37:465-468. doi:
- Tindall BJ, Grimont PAD, Garrity GM, Euzeby JP, (2005). “Nomenclature and taxonomy of the genus Salmonella”, International Journal of Systematic Bacteriology, 55:521-524. doi:
- Bao Y, Wadden J, Erb-Downward JR, Ranjan P, Zhou W, McDonald TL, Mills RE, Boyle AP, Dickson RP, Blaauw D, Welch JD, (2021). “SquiggleNet: real-time, direct classification of nanopore signals”, Genome Biology, 22(1):298. doi:
- Hall MB, Wick RR, Judd LM, Nguyen AN, Steinig EJ, Xie O, Davies M, Seemann T, Stinear TP, Coin L, (2024). “Benchmarking reveals superiority of deep learning variant callers on bacterial nanopore sequence data”, Elife, 2024;13. doi:
- Takiguchi S, Takeuchi N, Shenshin V, Gines G, Genot AJ, Nivala J, Rondelez Y, Kawano R, (2025). “Harnessing DNA computing and nanopore decoding for practical applications: from informatics to microRNA-targeting diagnostics”, Chemical Society Reviews, 54(1):8-32. doi:
- Kim BY, Gellert HR, Church SH, Suvorov A, Anderson SS, Barmina O, Beskid SG, Comeault AA, Crown KN, Diamond SE, Dorus S, Fujichika T, Hemker JA, Hrcek J, Kankare M, Katoh T, Magnacca KN, Martin RA, Matsunaga T, Medeiros MJ, Miller DE, Pitnick S, Schiffer M, Simoni S, Steenwinkel TE, Syed ZA, Takahashi A, Wei KH, Yokoyama T, Eisen MB, Kopp A, Matute D, Obbard DJ, O’Grady PM, Price DK, Toda MJ, Werner T, Petrov DA, (2024). “Single-fly genome assemblies fill major phylogenomic gaps across the Drosophilidae Tree of Life”, PLoS Biology, 22(7):e3002697. Epub 20240718. doi:
- Open Access Government, January, 2025, pages 48-49
- Strasser BJ. Collecting, (2010). “Comparing, and computing sequences: the making of Margaret O. Dayhoff’s Atlas of Protein Sequence and Structure, 1954-1965”, Journal of the History of Biology, 43(4):623-660. doi:
- Palmblad M, Hoopmann MR, Dorfer V, (2025). “A Special Software Issue in Celebration of Margaret Dayhoff’s 100th Birthday”, Journal of Proteome Research, 24(3):977-978. doi:
- UniProt Consortium (2025). “UniProt: the Universal Protein Knowledgebase in 2025”, Nucleic Acids Research, 53(D1):D609-D17. doi:
- Cook H, Ussery DW, (2013). “Sigma factors in a thousand E. coli genomes”, Environmental Microbiology, 15(12):3121-3129. Epub 20130829. doi: