
David H Peyton at Portland State University outlines how research has discovered that heme is key to malaria drug development
The loss of the antimalarial drug chloroquine to drug resistance was among the most significant detriments to the antimalarial effort over the last half century. This is in part because chloroquine’s target is not a protein – but rather heme, the by-product of haemoglobin digestion by the parasite. This made the evolution of resistance very slow – even in what must be regarded the worst and chaotic of circumstances. Even today’s artemisinin combination therapies, that are the current standards of care, require a heme-targeting partner drug for the artemisinin. Many current malaria drug discovery projects are aimed at finding targets other than heme, but these often will usually lead to the evolution of drug resistance much quicker, rather than leading to a new heme-targeted drug.
The goal of our work has been to provide a drug superior to what chloroquine ever was, specifically by appending to the chloroquine structure another portion that inhibits Plasmodium falciparum chloroquine resistance transporter (PfCRT), the primary transporter whose mutation is responsible for the resistance to chloroquine and drugs like it. Exports of the drug from the parasite’s digestive vacuole (DV) means that the drug can’t build up to a sufficient concentration at the site of action to kill the parasite. The resulting hybrid drugs, including our lead molecule, were designed to be at least as potent and safe as chloroquine and could be less susceptible to the evolution of resistance. This is especially because it has been ‘tuned’ to have a shorter residence time in the blood at sub-therapeutic dose levels, and because they have never been available as a monotherapy in the regions of the world where malaria is found. Yet these new drugs would be fast-acting, potent, and very reticent to develop drug resistance. We call these new drugs, reversed chloroquine drugs (RCQ drugs).
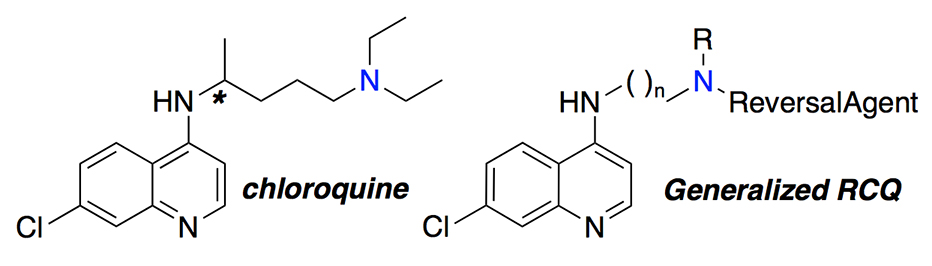
We tried the approach with a prototype RCQ drug a few years ago and then refined the structures to make a more practical drug in terms of what might make an orally dose-able medicine. We studied the mechanism of action of the RCQ drugs, to make sure that they were still working like chloroquine did, and to show that they were not very susceptible to resistance, both against strains of malaria from patients and evolved in the laboratory. This all gave rise to our lead molecule but the time and effort between deciding on a molecule and getting approved for a Phase 1 safety study (first-in-humans) are large, especially for an academic (university) laboratory.
Laboratory tests conducted for malaria drug development
There are also extensive studies required to demonstrate the likelihood of safety (non-poisoning) and a lack of genotoxicity (e.g., not carcinogenic) that are needed before a Phase 1 study is approved by a governing body, such as the Food and Drug Administration (FDA) in the U.S. These studies must be done under good laboratory practice conditions, which are generally available only from Contract Research Organizations (CROs), and not in-house at the university. In fact, a university researcher generally doesn’t even know what tests to do during this process, so consultants need to be hired. For these reasons, and others, I co-founded a start-up company, DesignMedix, Inc., which has the mission of discovering and developing drugs against infectious diseases, especially those which are evolving drug resistance. This began with malaria but now includes bacterial diseases.
We have therefore spent the last few years learning the process of bringing a candidate molecule from “Lead” to Phase 1 candidate. Last week we announced a contract with the National Institutes of Health/NIAID to carry out the Phase 1 first-in-human study. Of course, even a successful conclusion of this study will not be the end of this process: There will be at least one Phase 2 study that will be needed to be successfully completed, and then it will need to be partnered with another drug (and so, likely, another company) before the Phase 3 study (or studies). The entire process, from the start of the Phase 1 study, will likely be at least another 5 or 6 years before bringing a drug to market. That is if all goes well.
David H Peyton, PhD
Professor of Chemistry
Portland State University
Tel: +1 503 725-3875
peytond@pdx.edu
http://www.pdx.edu/chemistry/profile/dr-david-h-peyton
Please note: this is a commercial profile








