
Dr Gastone Castellani (biophysics) and Dr Jeanine Houwing-Duistermaat (statistics) from the University of Bologna, Italy develop methods for big multi-omics datasets
The aim of statistical analyses is to draw a conclusion for a population using data from a sample of this population. For example, we are interested in the relationship between cholesterol levels and body mass index (BMI), and we have data on these two variables in a sample. Using these data, we can compute the correlation between the two variables.
Statistical modelling
Under several assumptions, our calculated correlation, the estimate, is a good guess for the correlation between cholesterol and BMI in the population. If the sample size is large, then probably our estimate is close to the unknown true value. In statistics, methods are available to quantify the uncertainty of the estimator. Further, the sample needs to represent the population. This assumption might be violated when specific groups are underrepresented. For example, working mothers might not participate in the study, because they do not have time. Sometimes we can correct for such an underrepresentation, and the obtained estimate might still represent the parameter of interest.
Data quality
Many methods for data analyses also assume that the values in our spreadsheet are equal to the true biological value. In omics research, this is, however, typically not the case. Omics data can be of several types, such as genomic, metabolomic, proteomic and metagenomic and they are measured with various high throughput technologies. Now, several sources cause a discrepancy between the true and the observed value. A lot happens between taking the sample and measuring it in the laboratory. The sample might be transported and stored. The temperature and the time passing from sampling to measurement might affect the quality of the sample. Then the technology has a measurement error, which means that if we measure the same sample twice using the same machine at the same temperature, we will not get the same value. Finally, various technologies do not directly measure the amount of a molecule in the sample but base their value on a property of the molecule such as mass, which also leads to discrepancies between the observed and true values.
To draw conclusions using high throughput omics data from a sample for the population, it is important to treat all samples the same, minimize errors and use statistical modelling and methods which consider these errors. If the analysis method does not address the measurement error, the conclusion based on the sample for the population might be wrong and in contrast to what is often assumed the number of individuals cannot compensate for this error.
Privacy issues, federated learning, swarm learning and synthetic data generation
With big data measured on the same individual, privacy issues are becoming a concern. Moreover, international collaborations require building huge and shared databases across several countries. Among various methods for solving these issues, we can mention federated and swarm learning and synthetic data generation. Federated learning allows us to leave the data on local sites (for example hospitals) and to move the computational algorithms. After this step of “local learning,” there is a centralized phase of learning, usually by taking the average of the estimates of the model parameters. Swarm learning is a new technique that is becoming to be popular because it does not require the centralized phase of learning.
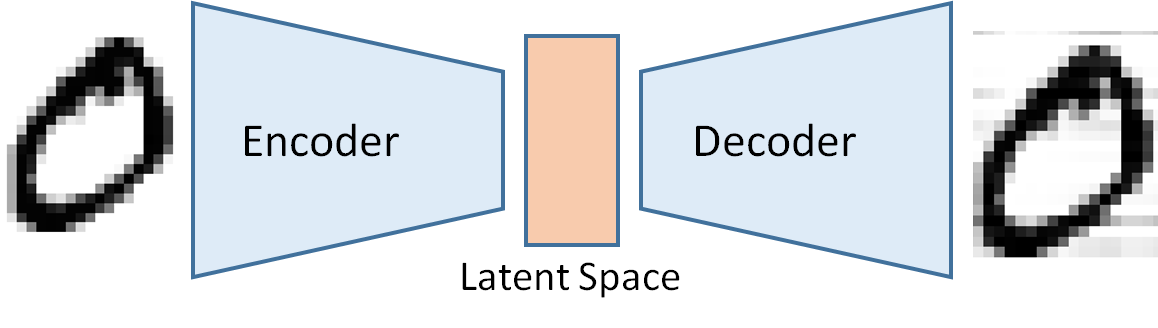
A synthetic data generation is an approach that not only leaves the data locally but also overcomes privacy issues. The basic idea of synthetic data generation is to sample in a latent space, which is constituted from variables that are not directly observable. Instead, they are transformations of the original observed variables. In mathematics, this is alled embedding on a low dimensional manifold or dimensionality reduction. This technique is extremely useful when we must deal with high-dimensional data sets. An example of a simple latent space is a line, where variables in this one-dimensional latent space are obtained by projection. The clue of this approach is that after embedding, it is possible to sample from this low dimensional manifold and reconstruct data that are like the original one in the larger space but not identical. The reconstructed data have the same statistical properties as the original data. The most celebrated techniques for synthetic data generation are the so-called variational autoencoder, which is composed of an encoder (the projection on the latent space) and a decoder (the reconstruction of pseudo- original data by sampling on the latent space).
Because omics datasets vary in properties such as scale, sparsity etc, the synthetic data generation process is heavily data-dependent. For example, genomic synthetic data can be produced by using generative Bayesian methods such as generative adversarial networks (GAN) and variational autoencoder (VAE) and can generate copy number variation, and genomic and chromosomal mutations. Current methods can generate next-generation sequencing data in a platform-dependent way: Illumina, Oxford Nanopore, PacBio etc. Metabolomics can be generated through a mixed approach based on stochastic simulation and probabilistic methods. Imaging data, such as magnetic resonance and X-ray based methods and histopathological whole slide imaging can be generated by using similar approaches (GANs, VAE) and other methods based on the encoder-decoder paradigm.
Synthetic data can also be used to adjust for the underrepresentation of certain groups in our sample. For example, we can generate additional gender and age-specific data if they are not sufficiently represented in our data set.
Thus, making valid conclusions using omics data requires sufficient knowledge about the sample, the measured dataset, and the available methods for storing, sharing and analysis of the data. This is only possible using an interdisciplinary approach.
This project has received funding from the European Union’s HORIZON 2020 Research programme under the Grant Agreement no. 101017549
Please note: This is a commercial profile.
© 2019. This work is licensed under CC-BY-NC-ND.








