
Dr James E. Goldman, Professor of Pathology and Cell Biology at Columbia University, describes gaining more insight into disease mechanisms by comparing mouse genetic models with the human disease when it comes to Huntington’s Disease
Huntington’s Disease (HD), a progressive neurodegenerative disorder, is characterised by involuntary movements, often with cognitive and psychiatric manifestations. It is caused by mutations in a single gene, HTT, which can be transmitted from parent to child. Usually, HD patients have one copy of the normal HTT and one copy of the mutant HTT (mHTT), so that a child’s chances of inheriting the mHTT from a parent are 50%.
In a previous Open Access Government article, two of us (Drs Osama Al Dalahmah and Goldman), describe a new technology, single nucleus RNA sequencing (snRNAseq), that has allowed us to look at the expression of genes in single-cells in HD brains. In comparing the genes that individual cells express in the HD brain to those in the brains of patients who do not have neurological disease, we found large numbers of changes. Some genes were expressed more highly, while others were expressed at much lower levels.
Huntington’s Disease research
We have examined different areas of the HD brains, including an area called the striatum, deep within the cerebral hemispheres. This area is markedly affected as HD progresses, suffering a loss of nerve cells, and accounting for the movement disorder. Other areas of the HD brain are also affected, such as the cerebral cortex, the outer layer of the brain, which helps to manage cognitive and emotional functions. We have found different changes in different areas of the HD brains.
Many human genetic diseases have been modelled in mice by adding the mutated human gene or fragments of the gene that causes the disease directly into the mouse DNA. These mouse models recapitulate many aspects of disease features that occur in humans. Genetic studies are easier in mice because their lifespan is much shorter than humans and we can modulate the disease through specific genetic and chemical treatments. Mouse models are useful in investigating the pathogenesis of a disease, for looking at the evolution of the disease over time, for understanding what drives disease, and for testing therapies. Although mouse models of disease show clinical and pathological similarities to the human disease, there are also often differences. This is not surprising, given the many differences between species. It is, therefore, very important to compare mouse models of diseases with their human counterparts. The snRNAseq technology has allowed us to investigate the comparison at a single-cell level. To do this, Drs Al Dalahmah and Goldman have been collaborating with Drs Leslie Thompson, Ryan Lim, and Jenny Wu at the University of California, Irvine.
Dr Thompson and her group have been working with a mouse model of HD. The R6/2 mice were generated by incorporating the first part of the mutated human HD gene into the mouse DNA. This piece of the mutant HTT gene is enough to cause a progressive neurological disease in which the mice experience involuntary, jerky movements and seizures (patients with juvenile, early-onset HD often experience seizures). The mice die prematurely.
Final remarks on HD
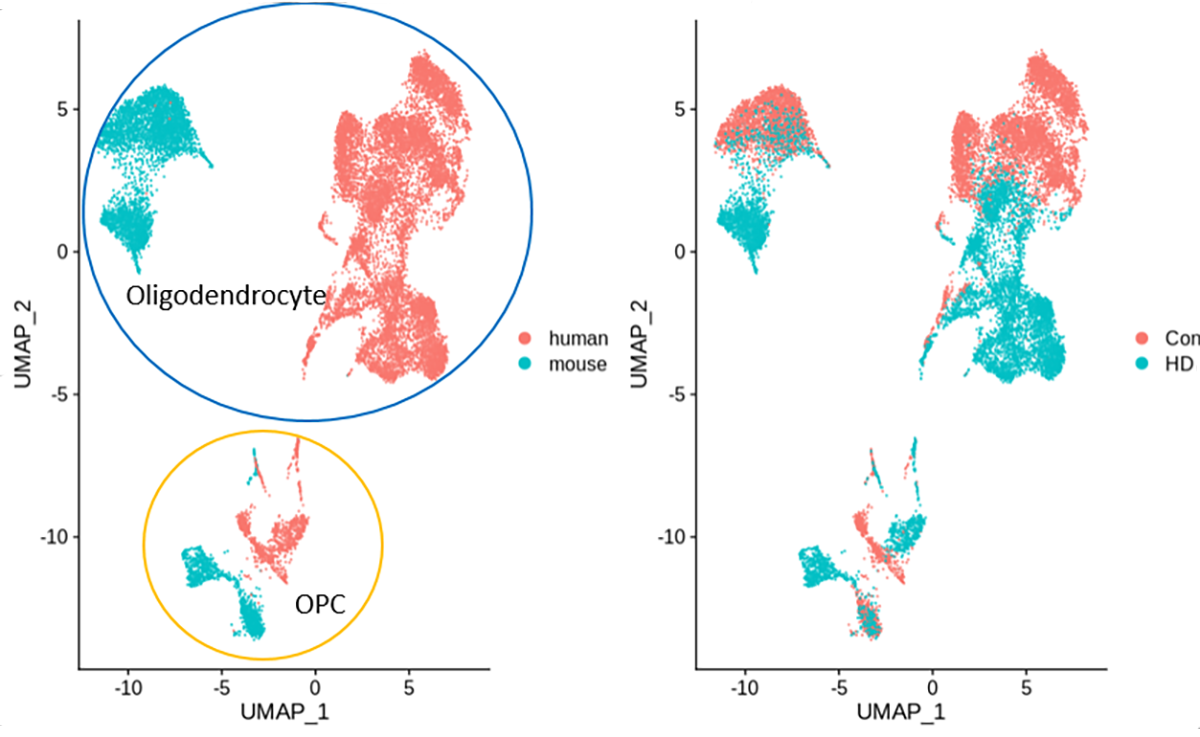
By using snRNAseq from our human HD brains and Dr Thompson’s HD mouse brains, we have been able to directly compare the changes in gene expression in all of the different central nervous system cell types. We have found many similarities in both neurons and glial cells between human and mouse HD, although there are some differences as well. As one example, we have found similar changes in oligodendrocytes, the cells that make myelin, the fatty insulating substance that wraps around the axonal processes of neurons, allowing for rapid conduction of information through the brain. Interestingly, both radiology and autopsy studies of patients with HD have discovered abnormally low myelination in the HD brain, and studies of mouse models have found similar pathology. Thus, our studies of single-cell gene expression in human brains and this mouse model helps to illuminate the basic molecular causes that underlie the myelination defects.
Furthermore, our observations allow us to understand in-depth how this particular mouse model replicates the human disease, a comparison that is extremely important in using mice to simulate human disorders. Now that we have identified that this important cell-specific feature of the disease occurs in both mouse and the human brain, we can use these mice to understand further how these cells become abnormal over time and test treatments that could lessen the overall disease.
Please note: This is a commercial profile






My husband was diagnosed with early stage Alzheimer’s and early stage Huntington’s disease. He is aware of what is going on some of the time but he refuses to bathe or do any of the things the doctors tell him. He could hardly get around because all he does is sit in front of the TV all day. He refuses to go for a walk or to a senior center and he refuses to bathe. I was beside myself as we cannot go anywhere when he is dirty and stinks. I didn’t know what to do, I could not physically overpower him and make him do things and when I ask him he tells me I am not his boss. There has been little if any progress in finding a reliable treatment. His Primary care provider introduced me to Kycuyu Health Clinic and their amazing Herbal treatments. The treatment is a miracle. the disease is totally under control. No case of delusion, disorientation, forgetfulness, making things up, hallucination, Muscle weakness, jumbled speech, loss of appetite or confusion in the evening hours.