")
Lalit P. Singh, Associate Professor at Wayne State University School of Medicine, explains the role of TXNIP in mitochondrial-lysosomal axis dysregulation in diabetic retinopathy
Diabetic retinopathy (DR) is a devastating disease affecting millions of people around the world, leading to blindness. Yet, there is no known cure till today. Diabetes is mainly of two types – Type 1 diabetes (insulin deficiency to due pancreatic beta cell death, an autoimmune disease, T1D) and Type 2 diabetes (insulin resistance, T2D) often associated with obesity. Whether it is T1D or T2D, sustained hyperglycaemia prevails in the blood and causes tissue injury.
Currently, the mechanism(s) for the initiation and progression of DR is not fully understood. One gene that is strongly induced by diabetes and high glucose in tissues, including pancreatic beta, renal and retinal cells, is the thioredoxin-interacting protein (TXNIP). TXNIP causes cellular oxidative stress, low-grade inflammation, cell death in DR.
TXNIP binds to thiroredoxin (Trx), inhibiting its anti-oxidant and thiol-reducing capacity. Trx1 is present in the cytosol/nucleus while Trx2 is in mitochondria (Fig. 1-I). Another cellular anti-oxidant system is the glutathione/glutathione peroxidase (GSH/GPX). However, under chronic hyperglycaemia, TXNIP continues to inhibit Trx1 and Trx2 causing cytosolic and MT reactive oxygen species (ROS) generation. Stressed mitochondria are inefficient in ATP synthesis while generating ROS. Therefore, removal of the damaged mitochondria is critical for cellular and MT health. For this, the damaged part of the mitochondrion is first separated by fission involving dynamin-related protein, DRP1 and fission protein, Fis1. Then, isolated mitochondria are removed by mitophagy (a specific process of autophagy) via lysosomal degradation.
The retina being a part of the central nervous system consumes large amounts of glucose and oxygen for its bioenergetics and visual function via the MT inner membrane electron transport chain (ETC). During ATP production, electrons leak, which are captured by O2-generating ROS. Although there are anti-oxidants in both the cytosol and the mitochondrion, they are overwhelmed by sustained blockade of the Trx-TrxR system by TXNIP. This leads to overutilisation of GSH.
Ultimately, MT damage occurs, which needs to be cleared by mitophagy, a complex process yet to be fully understood. Briefly, upon MT damage, PTEN-induced kinase 1 (Pink1) accumulates on the outer membrane and phosphorylates membrane proteins, which attract Parkin (an E3 ubiquitin ligase) marking for degradation (Fig. 1-II). TXNIP also interacts with REDD1 (regulated in development and DNA damage 1) and inhibits mTORC1 (mechanistic target of rapamycin complex 1), which phosphorylates ATG1 (ULK1) and ATG13 to block autophagy/mitophagy.
Furthermore, TXNIP/REDD1 inhibit ATG4B, which delipidates LC3B-II in autophagophore; thereby increasing double membrane autophagophore formation. Then, mitophagy receptors, optineurin and p62/sequestosome 1, are phosphorylated by TANK-binding kinase 1 (TBK1) enhancing their interaction with MT cargo and LC3B-II and mitophagophore formation.
The mitophagophore then fuses with lysosomes to form the autolysosome, which degrades the MT cargo. Disturbance of the lysosomal membrane by autophagy/mitophagy causes translocation of transcription factor EB (TFEB) to the nucleus and enhanced transcription of lysosomal and ATG genes and PGC1α, an MT biogenesis factor (Fig. 1-III). Although new mitochondria are generated, fusion with existing mitochondria to form functional MT network may be blunted due to MT stress.
Accumulation of fragmented mitochondria leads to bioenergetics deficiency. One aspect of TFEB regulation under mitophagy is that TFEB is phosphorylated by mTORC1 including at Serine 211 and sequesters in the cytosol via interaction with 14-3-3 scaffold protein while calcineurin-mediated dephosphorylation of TFEB leads to nuclear translocation and expression of CLEAR (Coordinated Lysosomal Expression and Regulation) genes. Which of the two proteins – mTOR or calcineurin – dominates the TFEB regulation in DR is yet to be defined?
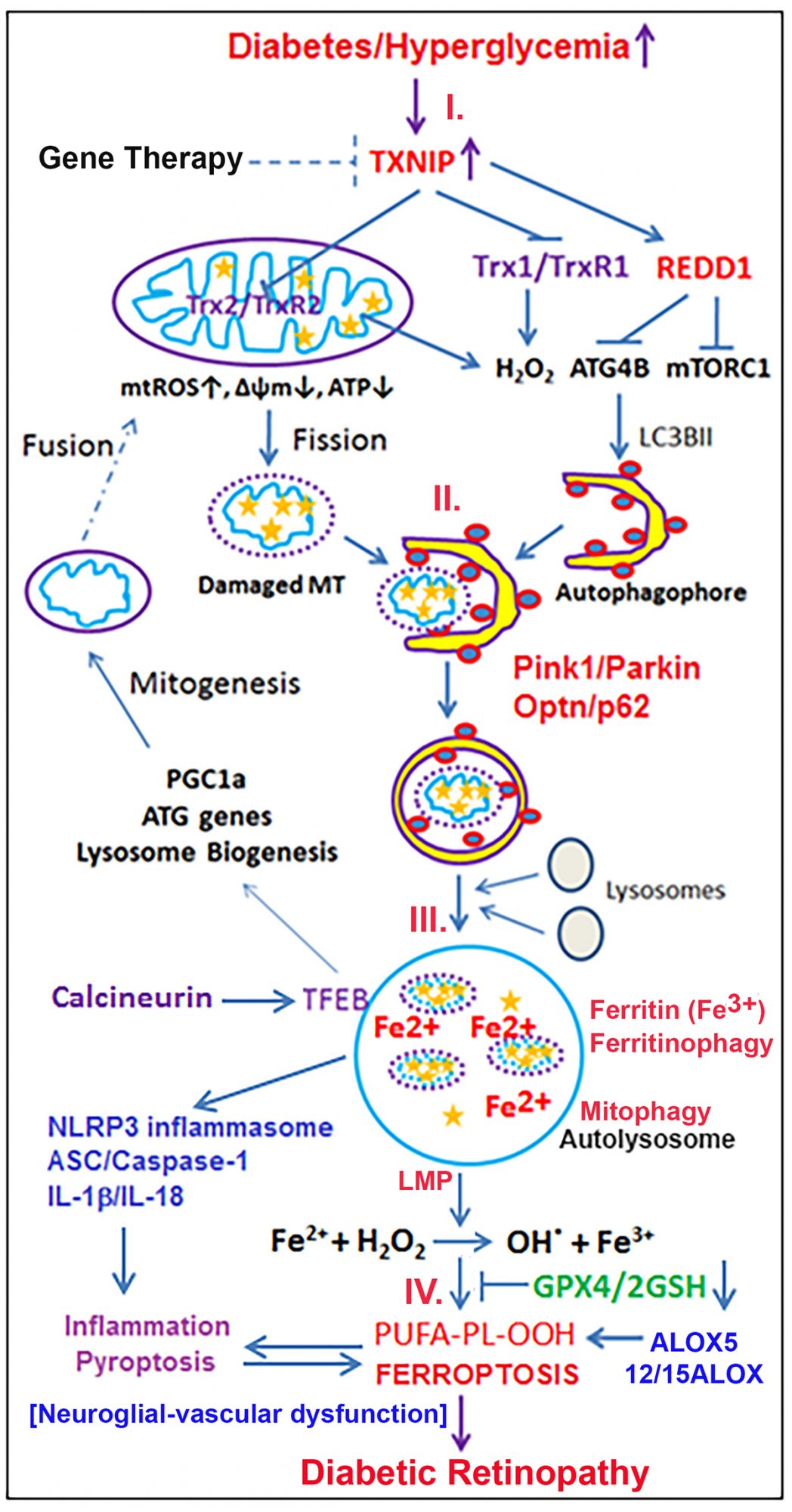
Another consequence of sustained oxidative stress and mitophagic flux in cells is the accumulation of damaged mitochondria, membrane lipids, oxidised proteins, free iron and H2O2 causing lysosomal enlargement, destabilisation and membrane permeabilisation (LMP) (Fig. 1-IV).
Mitochondria are major sites of iron metabolism including iron-sulphur cluster/complex biosynthesis, heme synthesis and storage in MT ferritin. MT TCA cycle enzyme (m-aconitase/ ACO2), Complexes I and III all contain 4Fe-4S clusters. In the cytosol, c-actonitase (ACO1) also contain 4Fe-4S cluster and serves as a dual function protein – (i) conversion of citrate to isocitrate and (ii) as an iron regulatory protein 1 (IRP1) in the absence of iron-sulphur. Under oxidative stress or hypoxia, 4Fe-4S complex in ACO1 is degraded and IRP1 binds to iron regulatory elements (IRE) in the 3’-UTR of transferrin receptor 1 (TfR1) mRNA and stabilises to increase TfR1 translation and iron uptake. Cytosolic iron is stored in ferritin (ferric iron, Fe3+) and iron utilisation requires autophagy of ferritin (ferritinophagy) to generate free/labile ferrous Fe2+, which is highly reactive with H2O2 (Fenton reaction) to generate reactive hydroxyl radicals (.OH) and ions (OH-).
Subsequently, oxidative stress (.OH) and iron overload cause plasma membrane phospholipid peroxidation (PL-OOH), mainly polyunsaturated fatty acids (PUFA) and ferroptosis, a non-apoptotic cell death mechanism by iron overloading and lipid peroxidation due to a decrease in the GPX4 activity. GPX4 is the sole enzyme that detoxifies PL-OOH using two GSH.
In addition, arachidonate 5-lipoxygenase (ALOX5) and 12/15ALOX, which are regulated by iron and oxidative stress, may also be activated and mediate PL-OOH.
Ferroptosis, being a non-apoptotic cell death, releases cellular DAMPs (damage-associated molecular patterns) including oxidised mtDNA and nuclear HMGB1 (high mobility group box 1), which further evoke innate immune responses.
Further, LMP activates NLRP3 inflammasome. ALOX5 generates leukotriene B4 (LTB4), which attracts immune cells such as microglia/macrophage mediating neuroinflammation and neurodegeneration in DR. Therefore, we propose that gene therapy for TXNIP knockdown and/or administration of mitochondria-targeted antioxidants (mito-Tempo, SS31), iron chelating agents (deferasirox, deferiprone) and ALOX inhibitor (Zileuton) may serve as potential combination therapies to prevent/slow down the progression of DR.
References
Perrone L, Devi TS, Hosoya K, Terasaki T, Singh LP. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol. 2009 Oct;221(1):262-72. PMID: 19562690.
Perrone L, Devi TS, Hosoya KI, Terasaki T, Singh LP. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis. 2010 Aug 19;1:e65. PMID: 21364670.
Singh LP. Thioredoxin Interacting Protein (TXNIP) and Pathogenesis of Diabetic Retinopathy. J Clin Exp Ophthalmol. 2013 Aug 5;4. PMID: 243539009.
Devi TS, Somayajulu M, Kowluru RA, Singh LP. TXNIP regulates mitophagy in retinal Müller cells under high-glucose conditions: implications for diabetic retinopathy. Cell Death Dis. 2017 May 11;8(5):e2777. PMID: 28492550.
Lalit PS, Thangal Y, Fayi Y, Takhellambam SD. Potentials of Gene Therapy for Diabetic Retinopathy: The Use of Nucleic Acid Constructs Containing a TXNIP Promoter. Open Access J Ophthalmol. 2018;3(2). PMID: 31106306.
Devi TS, Yumnamcha T, Yao F, Somayajulu M, Kowluru RA, Singh LP. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol Open. 2019 Apr 25;8(4). PMID: 31023645.
Funding:
NIH/NEI R01 EY023992 (LSP)
NIH/NEI core grant P30EY004068 (OVAS)
Research to Prevent Blindness (OVAS)
Lalit Singh Pukhrambam, PhD
Associate Professor
(Tenured) Department of Ophthalmology,
Visual and Anatomical Sciences (OVAS), Wayne State University School of Medicine
540 E Canfield, Scott Hall 8332
Detroit, MI 48201, USA
Tel: + 1 313 577 5032
ak1157@wayne.edu
www.anatomy.med.wayne.edu/profile/ak1157
*Please note: This is a commercial profile








