
Here, we learn about Daisuke Kihara, Professor of Biological Sciences and Computer Science at Purdue University, who develops state-of-the-art computational methods for modelling 3D structures of protein complexes
Cell: Network of protein-protein interactions for performing biological functions
Proteins are key working molecules and building blocks in a cell. They are involved in almost all functional activities within and between cells, which include metabolic and signalling pathways, gene expression, cell duplication, diseases, and viral infection. Proteins carry out these functions through physical interactions with other proteins and different molecules, such as DNA and RNA.
Therefore, determining the three-dimensional (3D) structures of protein interactions (complexes) is crucial for a molecule-level understanding of how proteins function.
The 3D structure of a protein complex also enables the development of drugs that interfere with interactions between proteins. Protein structures are primarily determined by experimental methods, such as X-ray crystallo-graphy and cryo-electron microscopy. However, many protein complexes are difficult to determine via experimental methods.
Computational protein docking
To compliment experimental app-roaches, many computational methods for protein docking have been developed, which build 3D models of protein complexes by assembling component protein structures. The importance of protein docking has been recently highlighted due to the increasing number of experimentally solved monomeric structures, as well as recent computational methods, such as AlphaFold, can now predict monomeric protein structures with a high degree of accuracy. Thus, protein docking has become the frontline of computational biology and systems biology.
The performance of protein docking methods has been periodically evaluated by the community-wide “Critical Assessment for Protein Interactions” (CAPRI) competition. In CAPRI, Dr Kihara’s protein docking methods have been routinely ranked among the top of all groups. Here, we introduce a series of protein docking methods developed by Dr Kihara’s group.
The LZerD protein docking software suite
Many protein complexes in a cell are formed between two proteins (pairwise docking). Dr Kihara has developed a pairwise docking method, LZerD (Local 3D Zernike Descriptor-based protein docking). LZerD is unique among other comparable methods in that it uses a mathematical molecular surface representation, termed a 3D Zernike Descriptor, which allows soft interactions between proteins, which mimics the interactions of real proteins. After the successful development of LZerD, Dr Kihara’s team has further extended LZerD’s capabilities in several directions.
In a cell, there also exist large protein complexes consisting of many more than two proteins. Such complexes with multiple proteins can be modelled with the Multi-LZerD method. To build a multi-protein complex, Multi-LZerD starts by generating a pool of pairwise docking models of pairs of component proteins. Then, the pairwise models are used as building blocks and combined to construct full-component complexes. Using Multi-LZerD, the assembly order of individual proteins can also be identified by observing the energetics of complex assembly pathways in the modelling process. This assembly order prediction has been made available in the Path-LZerD program.
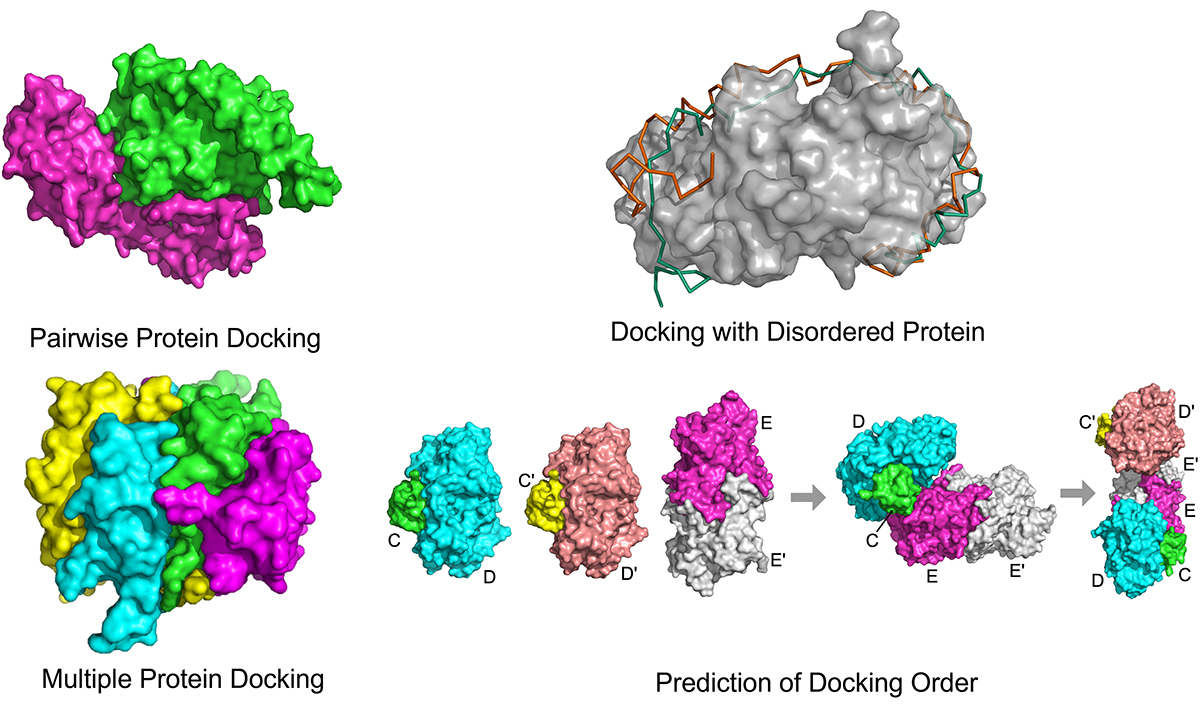
A typical protein stays folded in a stable, globular shape. However, it has been noticed that there are a substantial number of proteins, including over 20% of proteins in humans, that are naturally disordered – which means that such proteins, termed intrinsically disordered proteins (IDPs), are inherently flexible and do not fold into a particular shape. IDPs tend to interact with many proteins, taking advantage of their flexible nature. To consider IDPs in computational protein docking, Dr Kihara has developed IDP-LZerD, which can dock an IDP to a folded protein by first docking small fragments of the IDP, which are later connected together.
Altogether, the LZerD docking suite has significantly expanded the applicability of computational protein docking to address different types of macromolecular modelling.
Docking meets artificial intelligence
Artificial intelligence, particularly machine learning, has revolutionised many research areas, including image processing and language translation, and computational biology is not an exception. Dr Kihara’s group has used deep learning, a powerful machine learning method, in the protein docking field. In the methods they have developed, they consider 3D models of protein complexes as 3D images or graphs, and developed deep-learning-based methods that can distinguish correctly docked models from incorrectly docked ones.
Providing easy use of the software to the scientific community
Protein docking methods developed by Dr Kihara’s group are freely available to use via a web-server, https://lzerd.kiharalab.org. The software is also provided as a standalone package or source code.
The future: Computational simulation of a whole-cell
The protein docking methods developed by Dr Kihara’s group build the individual interacting units that make up entire cellular protein-protein interaction networks. The computational biology field is moving quickly. By combining other techniques, such as physical simulation of proteins and other molecules, Dr Kihara foresees a near future that a cell-scale dynamic simulation of protein interactions will be possible in silico.
For more information, please visit: https://kiharalab.org/
References
Aderinwale T et al., Computational structure modeling for diverse categories of macromolecular interactions. Curr. Opin. Struct. Biol., 64: 1-8 (2020).
Christoffer C et al., LZerD webserver for pairwise and multiple protein-protein docking. Nucleic Acid Research 49: W359-W365, (2021).
Peterson LX et al., Modeling disordered protein interactions from biophysical principles. PLoS Computational Biol., 13: e1005485, (2017).
Peterson LX et al., Modeling the assembly order of multimeric heteroprotein complexes. PLoS Comput. Biol., 14: e1005937 (2018).
Wang X. et al., Protein docking model evaluation by graph neural networks. Frontiers in Molecular Biosciences, 8: 647915 (2021).
Please note: This is a commercial profile
© 2019. This work is licensed under a CC BY 4.0 license.








