
Dr. Arun Srivastava, George H. Kitzman Professor of Genetics, from the University of Florida describes the rationale and strategies for the development of next generation (NextGen), generation X (GenX) and optimised (Opt) recombinant AAV vectors for human gene therapy
Recombinant AAV vectors have been, or are currently being, used in 165 Phase I/II clinical trials (and one recent Phase III trial) in humans and thus far, no serious adverse events have ever been observed or reported. AAV2 vectors have shown clinical efficacy in three human diseases: Leber’s congenital amaurosis (LCA), aromatic L-amino acid decarboxylase deficiency (AADC) and choroideremia. In the past decade, at least 12 additional AAV serotype vectors have also become available. AAV1 vectors have successfully been used in the gene therapy of lipoprotein lipase deficiency (LPL) and α1-antitypsin deficiency (AATD); and AAV8 vectors have shown clinical efficacy in haemophilia B; AAV5 vectors in haemophilia A; and AAV9 vectors in spinal muscular atrophy (SMA) and Pompe disease. The AAV1-LPL vector was approved as a drug, designated as Alipogene tiparvovec and marketed under the trade name Glybera®, in Europe in 2012.
In 2017, an AAV2 vector expressing retinal pigment epithelium-specific 65 kDa protein (RPE65) was licensed as a drug, Voretigene Neparvovec (Luxturna), in the USA. Additional Phase I/II clinical trials are currently being pursued with AAV1, AAV2, AAV3, AAV5, AAV6, AAV8, AAV9 and AAV10 vectors for the potential gene therapy of a wide variety of human diseases.
Despite these remarkable achievements, it has become increasingly clear that the full potential of AAV vectors composed of the naturally occurring, wild-type (WT) capsids and genomes is unlikely to be realised for the reasons outlined below.
AAV capsid is targeted for degradation by the host cell enzymes
Since the wild-type [WT] AAV did not evolve for the purposes of delivery of therapeutic genes, recombinant AAV vectors composed of naturally occurring capsid are unlikely to be optimal in human clinical trials. Dr. Srivastava’s laboratory made the original observation that following infection of cells, only ~20% of the input AAV2 vectors gain entry into the nucleus, whereas ~80% fail to escape the endosome in the cytoplasm. It was subsequently reported that AAV2 capsids become ubiquitinated in the cytoplasm, where they are targeted for degradation by the host cell proteasomal machinery, thereby negatively impacting the transduction efficiency of the first generation of AAV vectors.
The WT AAV2 capsid contains specific amino acid residues (7 tyrosines {Y}, 17 serines {S} and 15 threonines {T} that are surface-exposed and are targeted for phosphorylation by the host cell protein kinases. In addition, AAV2 capsid also contains seven surface-exposed lysine (K) residues, which are ubiquitinated. These modifications render a large fraction of first generation of AAV vectors for degradation by the host cell proteasome machinery. Each of these surface-exposed Y, S, T and K residues were mutagenised in Dr. Srivastava’s laboratory to generate the next generation (NextGen) of AAV vectors with improved intracellular trafficking to the nucleus and consequently, high-efficiency transduction.
Interestingly, most, if not all, of the surface-exposed Y, S, T and K residues are highly conserved among all 10 commonly used AAV serotype vectors and most of these residues have also been mutagenised in each of the 10 AAV serotype vectors. Thus, the capsid-modified next generation of AAV vectors, as schematically illustrated in figure 1, overcome some of the limitations associated with the first generation of AAV vectors.
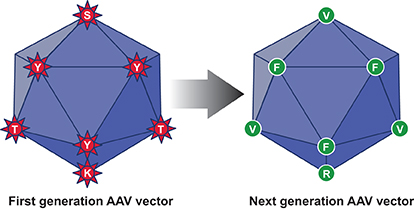
In this context, it is important to point out that three Phase I/II clinical trials with one of these tyrosine-mutant AAV2 vectors have been initiated and the initial results appear very promising. In one trial, 13 of 14 patients with Leber’s hereditary optic neuropathy (LHON), who were administered this vector intravitreally, showed improvement in visual acuity up to 24-month follow-up, without any loss of vision or any serious adverse events. Thus, the capsid-modified next generation of AAV serotype vectors will prove to be far more efficacious than their WT counterparts in human gene therapy.
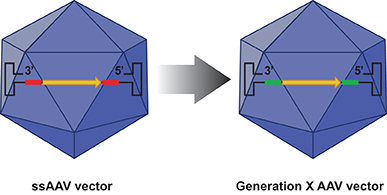
Figure 2. The D-sequence in the ssAAV genome (depicted in red) is replaced by a substitute sequence (depicted in green) in the generation X (GenX) AAV vectors that mediate more efficient transgene expression. Encapsidation of the GenX genome into the NextGen capsid leads to the generation of more efficient optimised (Opt) AAV vectors.
The single-stranded AAV genome is transcriptionally-inactive
The WT AAV genome is a single-stranded DNA and single-stranded DNA of both [+] and [-] polarities are encapsidated into separate mature virions with equal frequency. While inconsequential for the WT AAV, which remains latent in host cells, the single-stranded DNA of a therapeutic gene in an AAV vector is problematic, since single-stranded DNA is transcriptionally-inactive and viral second strand-DNA synthesis is a rate-limiting step during AAV vector-mediated transgene expression.
Dr. Srivastava’s laboratory identified that FKBP52, a well-known celliular chaperone protein that binds the immunosuppressant drug FK506, interacts specifically with the single-stranded sequence of 20 nucleotides, termed the D-sequence, within the AAV inverted terminal repeat (ITR) at the 3’ -end of the viral genome and strongly inhibits the viral second-strand DNA synthesis, resulting in impaired transgene expression. In nearly all clinical trials performed thus far have utilised ssAAV vectors and yet clinical efficacy has been observed in several human diseases, as outlined above.
Dr. Srivastava hypothesised that substitution of the D-sequences from the viral genome would allow more efficient viral second-strand DNA synthesis and consequently high-efficiency transgene expression. However, both D-sequence-substituted AAV genomes failed to undergo encapsidation. Interestingly, when only one of the two D-sequences was substituted from the AAV genome, successful encapsidation ensued and the efficiency of transgene expression from the D-sequence-substituted AAV genomes was significantly higher than that from their unmodified counterparts. These genome-modified vectors, designated generation X (GenX) AAV vectors are depicted schematically in figure 2.
Further studies revealed that the D-sequence at the 5’ -end in the viral ITR contains the binding site for a cellular NF-κB repressing factor (NRF), which inhibits viral transgene expression. Thus, the genome-modified GenX AAV vectors partially overcome the limitation associated with the conventional ssAAV vectors.
More recent studies from Dr. Srivastava’s laboratory have documented that when the modified AAV genomes are encapsidated into the modified capsids, the resulting optimised (Opt) AAV vectors transduce cells and tissues significantly more efficiently at 20-30–fold further reduced doses. Thus, the NextGen, GenX and Opt AAV serotype vectors circumvent the problems associated with the first generation of AAV vectors, in addition to being far more efficacious, less immunogenic and more cost-effective.
Plans are currently underway to use one of these NextGen AAV vectors to initiate a Phase I clinical trial in humans in a three-way collaboration among Christiann Medical College in India, and Emory University and University of Florida in the USA.
Please note: this is a commercial profile
Dr. Arun Srivastava PhD
George H. Kitzman Professor of Genetics
University of Florida
Tel: +1 352 273 8259
http://research.pediatrics.med.ufl.edu/researchers/research-faculty/arun-srivastava







")
