
Michael Roth, Research Group Leader at University Hospital Basel asks if fine dust air pollution (PM2.5) is a cause of chronic inflammatory lung diseases and provides a most engaging response
Chronic airway inflammatory diseases including asthma, chronic obstructive pulmonary disease (COPD) and lung fibrosis have been increasing worldwide for several decades. The initiation of asthma is often seen as an allergic over-reaction, however, over 45% of the patients are not linked to an allergic response. The cause of lung fibrotic diseases is largely unknown, but recent studies indicate that fine dust might be involved in the pathogenesis.
Regarding COPD, the cause seems to be clear: cigarette smoking, while the mechanism by which inhaled cigarette smoke starts the pathogenesis of COPD is not well understood. Fine dust air pollution with particulate matter < 2.5 micron (PM2.5) has been recognised to contribute to the progression of all three chronic inflammatory lung diseases. Thus, one could question is if it should be considered as a major cause.
Particulate matter < 2.5 micron (PM2.5)
PM2.5 air pollution is a serious global health problem, especially in the extending urban areas of the ever-growing major cities. PM2.5 air pollution originates from open cooking fires (gas, petrol, wood), traffic, industry and agriculture. Moreover, PM2.5 is also a major component of cigarette smoke and it can easily pass through the cigarette filters.
Several studies showed that exposure to PM2.5 increased the prevalence of chronic inflammatory diseases of the immune system, the lung, the cardiovascular and gastrointestinal organs. A new threat is presented by fine plastic particles that can enter the human food chain through fish and seafood. It was thought that the major problem with such particles was their toxicity, which varies with their chemical composition. However, new studies indicated that the real problem is their tiny size. For most organisms, it is difficult to get rid – excrete – such small particles once they are in the body, regardless if they were inhaled, ingested or taken up by other ways.
Regarding the lung, it has been reported that repeated exposure to PM2.5 accelerated the decline of lung function, caused irreversible airway wall remodelling and increased the exacerbation rate in asthma patients. Even in the healthy lung, PM2.5 increased oxidative stress and the expression of pro-inflammatory cytokines and growth factors, which had been associated with asthma, COPD and fibrosis.
What is more worrying, are the observations that exposure to PM2.5 during embryogenesis and during early childhood (years 0-6) predisposes the lungs to develop chronic inflammatory lung diseases later in life. The molecular biological mechanism(s) by which PM2.5 leads to chronic lung inflammation remains largely unknown (Fig. 1). Toxicity due to the chemical nature of PM2.5 is one aspect of their action, but this applies mainly to soluble particles.
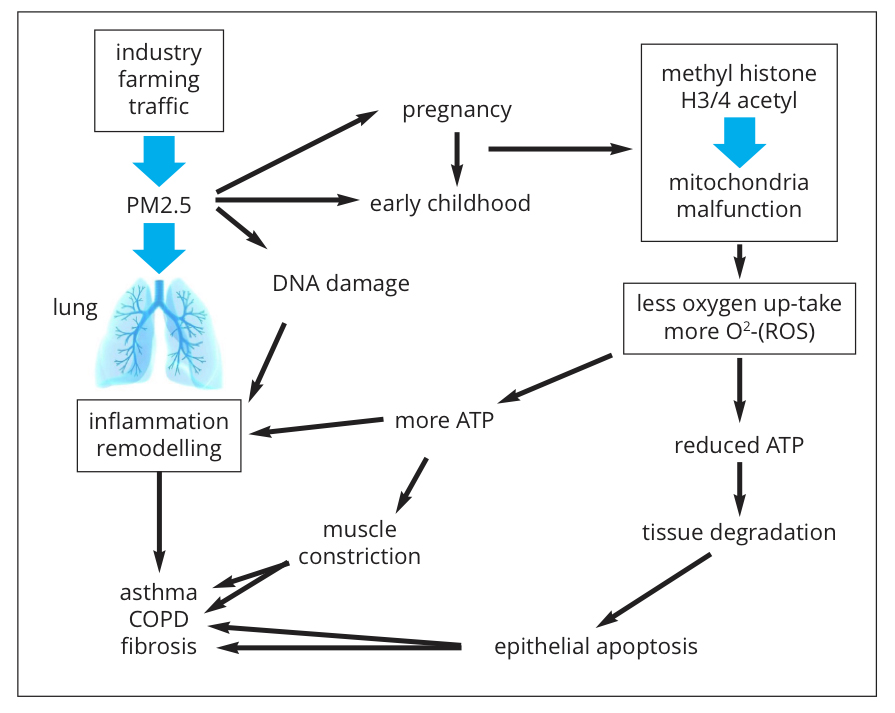
The size of insoluble PM2.5 compounds is the biggest concern. These small particles can be taken up not only by specialised immune defence cells, such as macrophages but also by tissue forming cells (muscle cells, connective tissue cells, etc.). Once inside a cell, the small particles activate an inflammation-like response, which aims to get rid of them, but which is most often not successful (Fig. 1).
Inhaled PM2.5 is deposed in the mucosa, causing epithelial cell damage, a typical pathology of asthma and COPD. PM2.5 stimulated the release of inflammatory mediators and lowered the threshold of allergic responsiveness, thus, they may increase the frequency of exacerbation in asthma and COPD. Moreover, some reports indicated that the effect of inhaled PM2.5 are different when they are deposed in the larger airways, compared to in the smaller airways, or in the alveoli where the gas exchange (O2 exchange for CO2) takes place. Thus, understanding the cell and molecular biological effects of insoluble PM2.5 particles seems to be crucial if we want to find therapies or cures for the diseases they cause.
Increasing evidence shows that PM2.5 can induce epigenetic modifications that regulate the gene activity on multiple levels, such as histone and DNA structure. Earlier, I reported that epigenetic events may underlie the “inheritable” predisposition of the lung to chronic inflammatory diseases. In this regard, mitochondria have to be considered to play a central role. The mitochondria are so-called organelles and are partly independent of the cells function and they have their own DNA and specific proteins. Mitochondria are the cell’s power stations and their function is essential for life. Modified mitochondria activity was linked to metabolic diseases, inflammation, cancer and tissue degeneration. PM2.5 can modify the function of mitochondria.
PM2.5 affected the function of mitochondria and the endoplasmic reticulum. In animal models, PM2.5 induced (i) endoplasmic reticulum stress, (ii) disturbed embryonal development and (iii) initiated the formation of fibrotic lesions in other organs other than the lung. In more details, Akt – mTOR signaling regulated mitochondria activity through mitochondria-associated endoplasmic reticulum membranes.
This observation linked PM2.5 to our own studies on Akt – mTOR signaling as a central regulator of mitochondria function which controls tissue remodelling in chronic lung inflammation. In allergic asthma, we showed that IGE over-activated the inflammatory Akt – mTOR signaling by down-regulating its inhibitor PTEN, which controls also the interaction between mitochondria and the endoplasmic reticulum. This suggests that the interaction between mitochondria and the endoplasmic reticulum is part of the pathogenesis of asthma and may also be relevant to other chronic inflammatory lung diseases.
The endoplasmic reticulum is closely linked to mitochondria function and together, they regulate protein synthesis, protein folding and cellular calcium metabolism. The balance between these mechanisms is crucial to prevent chronic lung inflammation. The control of hormone receptors and histones is central to the effect of drugs used in the therapy of chronic inflammatory diseases.
What is known about the effects of PM2.5 on mitochondria?
In rats, inhaled PM2.5 increased the formation of radical oxygen species (ROS) which caused the dysfunction of mitochondria. In other studies, PM2.5 reduced the oxygen consumption of mitochondrial oxygen, thereby, it decreased ATP production and caused mitochondria fragmentation. The result of this reduced energy production by mitochondria was cell death, which may be linked to the decay of the lung tissue which often occurs during the end-stage of COPD and is called emphysema. Regarding COPD, PM2.5 contained in cigarette smoke modified the methylation pattern of DNA and proteins also in non-smokers.
Concerning asthma, increased death of epithelial cells by PM2.5 exposure might explain the disrupted structure of the epithelium that lines the bronchus and is the barrier for inhaled dust, bacteria and viruses. PM2.5 caused histone hyper-methylation, thereby, modifying the accessibility of genes for transcription proteins. What is particularly worrying is the observation that such a process could happen during embryogenesis and the effects were lasting into childhood.
In animal models, exposure to PM2.5 early in life induced epigenetic predisposition to asthma-like symptoms such as, increased oxidative stress and extracellular matrix remodelling; which were handed down to the next generations. Particles that are < PM2.5, – PM0.1 –, have even been found accumulated inside mitochondria and caused severe damage to the mitochondrial DNA. Unfortunately, only a small number of studies have ever attempted to investigate this effect of small particles.
Mitochondria activity was up-regulated in asthmatic mesenchymal cells through the protein methylation catalysing protein PRMT1. However, in COPD, the same mechanisms caused the deterioration of mitochondria. In other animal models, PRMT1 was the major regulatory enzyme for mitochondrial proteins. It has not been investigated if the same mechanism is relevant to the pathogenesis of chronic inflammatory lung diseases in humans.
Conclusion
We need to find new therapies. For asthma alone, the number of patients doubles every decade and affects 350,000,000 people worldwide at present. The facts that (i) Exposure to PM2.5 during late embryogenesis and repeated exposure during early childhood predisposes the lungs to develop chronic inflammation later in life; (ii) PM2.5 induces epigenetic modifications of proteins and DNA and; (iii) These epigenetics can be handed down over generations – strongly suggest that we need to understand this mechanism(s) when we want to find new therapies and cures for chronic inflammatory lung diseases.
Please note: This is a commercial profile





