
Professor Olivier Braissant highlights here, how the challenges of treating genetic diseases can be exemplified by research on creatine transporter deficiency
Treating genetic diseases has always been a challenge. A relatively small number of these diseases can be treated through specific diet, medics, the use of chaperone molecules or enzyme replacement therapies. A few recent advances also brought gene therapy to the panel of treatments. However, the vast majority of genetic diseases remains, so far, beyond possibilities of treatment, and research activities continue to be hardly needed with the goal to offer therapies to the affected patients.
Cerebral creatine deficiency syndrome
Cerebral creatine deficiency syndromes (CCDS) represent good examples of this difference between treatable and so far, untreatable genetic diseases. CCDS are caused by mutations occurring in the genes coding for arginine: glycine amidino transferase (AGAT) and guanidinoacetate methyltransferase (GAMT) (the two enzymes of the endogenous creatine synthetic pathway), as well as for the creatine transporter (SLC6A8 or CRT). These three genetic diseases are characterised by a lack of, or very strong decrease in creatine (Cr) in the brain when measured by proton magnetic spectroscopy (1H-MRS), and lead to severe neurological symptoms like intellectual disabilities, defects in speech acquisition and in some cases refractory epilepsies or autism spectrum. CCDS account for 1-2% of all mental retardations, Cr transporter deficiency (CTD) being the most frequent (1% of all mental retardations in boys, the SLC6A8 gene being X-linked). Cr allows the regeneration of ATP and the cellular storage of high energy phosphates under its phosphorylated phosphocreatine (PCr) form (by the Cr/PCr/creatine kinase system), is one of the main osmolytes of the brain, and was also recently suggested as a neuromodulator or neurotransmitter.
Creatine
AGAT and GAMT deficiencies can be treated by Cr supplementation, allowing the slow but significant replenishment of the patient’s brain with Cr, and the improvement of their conditions (the possibility of a pre-symptomatic treatment even allowing the normal brain development of the concerned children).
In contrast, despite more than 20 years of research since its discovery, SLC6A8 deficiency has so far remained refractory to any efficient treatment. Cr supplementation has been tried without any success to replenish the brain with Cr. The reason for this is that SLC6A8 is the sole way for Cr to enter the brain from the periphery at the blood-brain barrier (BBB). Cr precursors arginine and glycine, as well as Cr derivatives being transported independently from SLC6A8, have been tried also but without any success either.
Creatine transporter deficiency is the archetype of a genetic disease being extremely challenging to treat. The disease essentially affects the brain, and the only way to treat it means being able to restore reasonable levels of Cr (or Cr derivatives performing the same functions as Cr) within the brain tissue. However, making a molecule enter the brain from the periphery is difficult. To reach neurons or oligodendrocytes (the cells making myelin sheath in the white matter), a molecule coming from the blood must first cross the microcapillary endothelial cells at BBB (one uptake and one efflux mechanisms), secondly cross the astrocytes surrounding BBB (another uptake and another efflux mechanisms), to finally reach the target cell through a third uptake mechanism (thus in total five membrane crossings).
However, new hopes for the treatment of SLC6A8 deficiency are arising nowadays, with the development and availability of new chaperone molecules and delivery routes, as well as new gene therapy technologies.
Cr transporter deficiency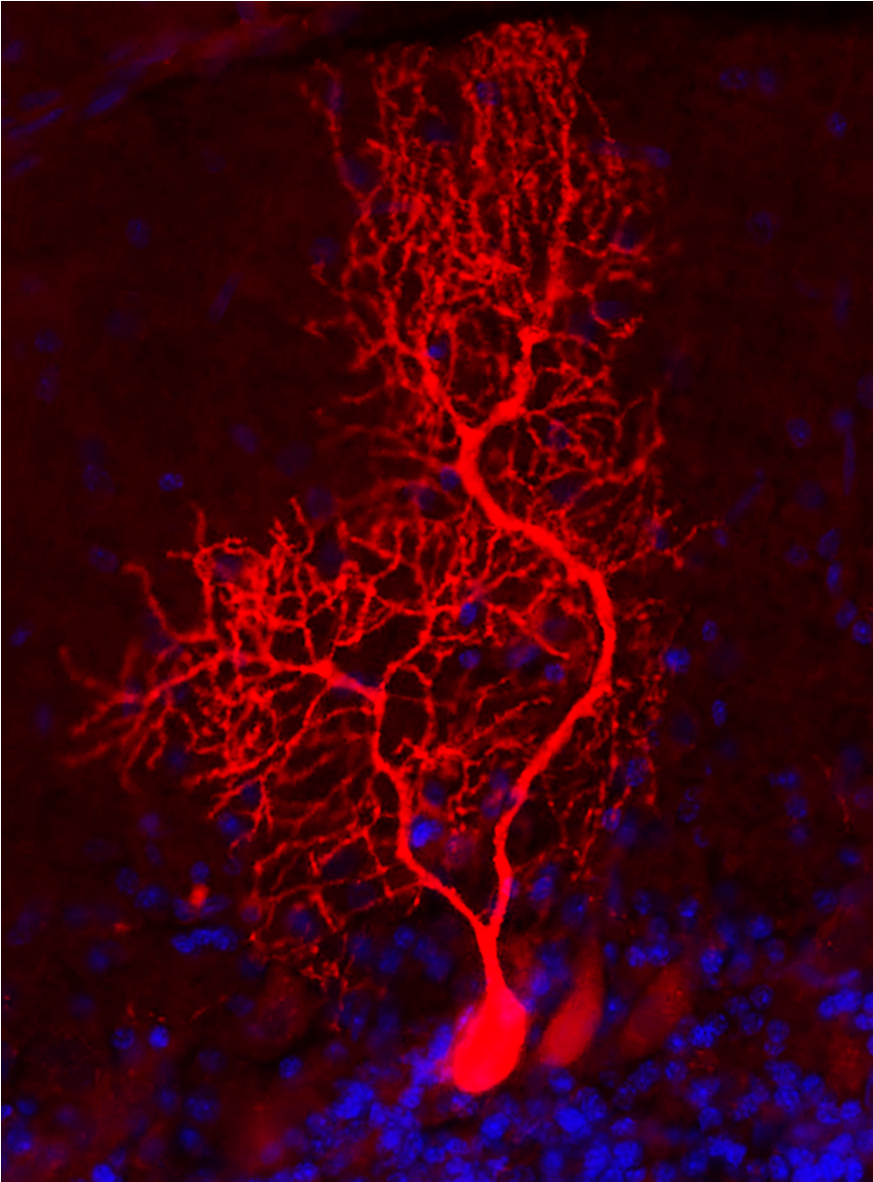
For CTD caused by missense mutations (e.g., mutations causing the change in one amino acid in the protein sequence, with the gene continuing to be expressed but the protein-losing part or totality of its activity), a lot of research is performed to discover molecules that would work as chaperones. Chaperones are molecules (small molecules or proteins) that allow the correct folding of a misfolded protein (which often occurs in case of missense mutations). For CTD, these potential chaperones molecules would have to cross BBB and enter the brain cells where the misfolded SLC6A8 protein is expressed, help its correct folding and make the mutated creatine transporter regain its activity. Promising in vitro assays have already identified a few chaperone candidates to treat CTD, and research is now ongoing during in vivo trials.
Research to treat CTD is also performed to find new delivery routes to the brain. One of them is the nose-to-brain delivery route, for which a spray is applicated in the nasal cavity, delivering nanovesicles containing either Cr or Cr derivatives able to cross directly from the tip of the nasal epithelium into the brain (at the level of the olfactory bulbs) and then diffuse within the brain to deliver their content.
Adeno-associated viruses
Recently, the use of adeno-associated viruses (AAV) as vectors to treat genetic diseases by gene therapy has gained considerable attention, with already a few successes for genetic diseases affecting both peripheral tissues and nervous system (e.g., spinal muscular atrophy or adenosine deaminase severe combined immunodeficiency). As the brain remains challenging to treat, we and others are developing optimised AAV vectors able to cross BBB and efficiently transduce the brain tissue, with the aim of re-establishing the expression of a functional Cr transporter (SLC6A8) within the different brain cells normally expressing it (BBB, neurons, oligodendrocytes) and allowing the brain of CTD patients to recover their normal Cr level. The aim of our research is also to remain the simplest in our treatment way, e.g. developing vectors being injected intravenously in the patient and reaching his brain through normal circulation. We actually are in the first pre-clinical phase of this development, using a new in vivo rat model of CTD, the Slc6a8Y389C/y knock-in rat to demonstrate the proof of concept of our AAV strategy to treat CTD. Having already demonstrated that our AAV vectors efficiently transduce the desired brain cells (after intravenous as well as intra-cisternal injections, see Figure), we are in the process to perform our first injections of AAV vectors transducing the functional SLC6A8, with the hope to demonstrate in the following months that we can correct CTD by our protocol.
Please note: This is a commercial profile
© 2019. This work is licensed under CC-BY-NC-ND.





