
Professors Darren Griffin and Mike Bruford (Universities of Kent and Cardiff) discuss what is meant by a “whole genome sequence” and how it is revolutionising conservation efforts
The United Nations Convention on Biological Diversity’s Targets (2010) required signatory countries, by 2020, to minimise genetic erosion and safeguard genetic diversity. Generating genome sequences of farmed/domesticated animals, their wild relatives, cultivated plants and socio-economically/culturally valuable species is needed to progress this goal. The purpose is to revolutionise our understanding of biology and evolution, safeguard biodiversity and create new societal benefits.
A notable example is the sequencing of the SARS-COV2 genome rapidly led to the ability to test and contact trace. While whole-genome sequencing of a microorganism is straightforward, doing so for more complex (e.g. animal or plant) species is still a relatively new concept. In 2001 the first mammals (human and mouse) appeared, the first bird (chicken) in 2004, and the first endangered species (giant panda) in 2010. Since then, the genomes of innumerable microorganisms, animals, plants and fungi have been sequenced, at least to some degree.
Global loss of biodiversity is alarming and increasing. Efforts that result from genome sequencing are, however, showing real promise slowing this trend. Genomic information can revolutionise traditional conservation efforts and provide novel insights with real-world applications. Generation of quality genomic data and its subsequent analysis, interpretation and application, is however challenging and there is a gap between basic research and practical solutions for conservation managers.
Indeed, robust systems must put in place before genome sequence information can be useful, such as:
- A quality, and properly annotated reference genome.
- Resequencing of key individuals, and proper analysis of enough of them.
- Identification of genes relevant to reproductive fitness and viability.
- Functional assays to test the phenotypic consequences of genetic variants.
- Comparative data from other species, especially when experimenting on endangered species is ethically unacceptable.
- Analytical pipelines to achieve all of the above.
Genomic studies have discovered that ‘species are not an island’ as we previously supposed. Indeed, genetic exchange among species appears to be the rule, not the exception, and may help species adapt (e.g. as shown in recent studies of neotropical butterflies Heliconius). Species identification has genuine implications, where populations, protected by their “species label” can benefit from extensive conservation measures or, conversely, be ignored if they don’t have the right one. Moreover, analysis of multiple genomes across numerous groups highlighted the importance of prioritising conservation of organismal groups with an inherent genetic ability to adapt to their surroundings.
A summary of how good quality genome sequences expedite conservation with real practical solutions is given in the table. On the ‘rubbish in, rubbish out’ principle however, poorer quality information is less useful.
What makes a good genome sequence? “Chromosome-level assemblies”
Modern DNA sequencing technology makes it now not too hard nor expensive to create a poor-quality genome. Assigning large blocks of sequence to an overall genomic “map” of an organism can however be problematic and/or expensive. Sadly, most sequenced genomes do not have a good “map” with all genes ordered and assigned in their proper place on chromosomes. Chromosomes are lengths of DNA (analogous to continents and islands) and in every cell of the bodies of all organisms, they are organised in exactly the same way. Every species has its own unique pattern of organization, like the coloured blocks of a national flag.
For instance, three blocks (red, white, and blue) could depict the flag of the Netherlands, Luxembourg, Russia or the former Yugoslavia, depending on the organization. Without this, genome assemblies are little more than a “bag of DNA.” Our work, coupled with contemporary sequencing technologies, uses approaches to take the existing data and visualise directly blocks of sequence as they appear in their rightful place (see figure). We call a full description of this and organised genome a “chromosome level assembly.”
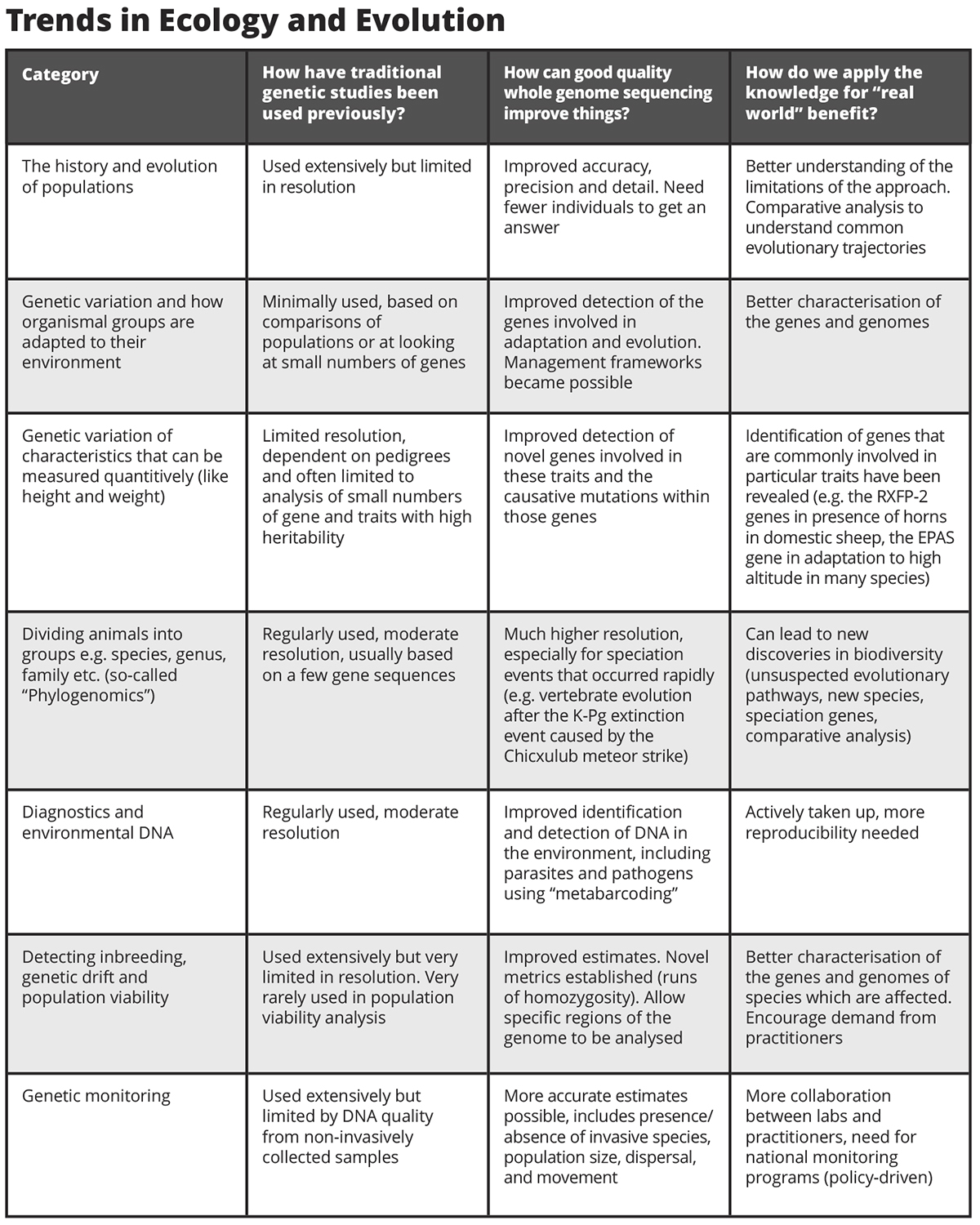
In agriculture, chromosome level assemblies can establish the relationship between the genome and the phenotypes of the plant or animal. They are used extensively in selection and breeding, leading to significant economic impact, more efficient food production and improved global food security. For conservation, with so many species on the CITES threatened/endangered list, such assemblies are essential. For example, some species (like Gyr and Saker falcons) inter-breed readily; hybrids and can be stronger and faster. Others like Peregrines do not form fertile hybrids, however. This is because of differing genome organization among species, something can readily be established with a chromosome level assembly.
At CryoArks we are dedicated to providing a sustainable genomic resource involving collection, curation and DNA extraction from numerous species worldwide. CryoArks aims to break down barriers to conservation management projects caused by permit problems by acquiring a large number of previously collected specimens, and sampling as many new species/populations as possible. Bringing these collections together, both physically and on databases, provides a resource that will be responsibly managed. We will ultimately achieve a step-change in the curation and availability of DNA, tissue and cell line samples, providing a forum for scientists and conservationists, with a self-sustaining resource, allowing conservation genomics to continue around the world.
*Please note: This is a commercial profile.








