
The annual worldwide cost of treating amyloid-associated diseases is about a trillion dollars and increasing steadily. But the human toll is worse
Amyloids are killing us, both literally and financially. Consider just these three: Amyloid β (Aβ) in Alzheimer’s Disease, α-Synuclein (α-Syn) in Parkinson’s Disease, and Amylin (aka IAPP) in Type 2 Diabetes. During my mother’s final decade, we could only try to make her comfortable as Alzheimer’s decimated her brain and stole her soul.
So why are treatments still so inadequate, especially for Alzheimer’s?
Dogmas, like Alzheimer’s patients, die slowly. Amyloid plaques composed primarily of amyloid beta (Aβ) fibrils and tangles composed of tau proteins are easily observed in the brains of most Alzheimer’s patients.
Decades of efforts to develop pharmaceuticals that target these large assemblies have failed to produce effective treatments or cures. Nonetheless, the media continues to assert that Alzheimer’s is caused by these hallmark structures. But they never mention that much smaller Aβ assemblies, called oligomers, are more toxic than fibrils (1), that these oligomers interact with cellular membranes to alter synapses (2) and to kill mitochondria (3) that power cells, or that the most toxic Aβ assemblies span membranes to form ion channels (4), or that specific ganglioside lipids and cholesterol are involved in channel formation, and Aβ-induced membrane dysfunction (5, 6), or that synaptic impairment is associated with entry of calcium into presynaptic terminals (7). These effects, which precede the formation of plaques and tangles, may be the root cause of the disease.
So why are oligomers and membrane permeation often ignored by the media and the pharmaceutical industry?
A common misconception is that amyloid oligomers are intrinsically disordered. If this is always true, then oligomers would not be good targets for drugs and there would be no motivation to solve their three-dimensional structures. But analyses of sequences from distantly related vertebrates are inconsistent with that perception: Aβ sequences from humans, snapping turtles, and goldfinches are identical, and Aβ from the bamboo shark has only one mutation and which is highly conservative.
Likewise, sequences of the first 62 residues of Α-Syn and β-Syn families are almost the same even though these subfamilies diverged long ago. Conserved portions of homologous proteins usually have similar highly ordered three-dimensional structures. Furthermore, portions of membrane-spanning assemblies exposed to lipid alkyl chains invariably have highly ordered structures. Highly conserved proteins typically perform important functions. Synuclein oligomers are involved in synaptic transmission and lipid transport (8); Aβ oligomers have antimicrobial (9) and other functional roles. However, the preservation of beneficial properties is often ignored in drug development.
If some oligomer-sized amyloid assemblies have highly ordered structures, then why haven’t they been solved?
Protein structural determination typically requires the isolation of a single type of structure in which proteins or subunits with identical sequences all have the same structure. A major impediment to amyloid structure determination is polymorphism.
My group has been developing structural models of non-fibril amyloid assemblies in different environments, namely the aqueous phase, lipoprotein complexes, and in cellular membranes (10, 11, 12, 13, 14). Our models are hypothetical because data have not been obtained that is precise enough to solve the structures directly. However, they have been developed to be consistent not only with microscopy images but with numerous other experimental results and with well-established molecular modelling criteria and methods. This article focuses primarily on Aβ42, but many of the concepts apply to α-Syn and amylin as well.
Aβ peptides are formed by enzymatic cleavage of a much larger amyloid-beta precursor protein. The length of Aβ peptides varies from 36 to 49 residues because the cleavage does not always occur at the same location. The most abundant form, Aβ40, has 40 amino acid residues, whereas the most toxic form, Aβ42, has two more at the end. Lipid bilayers are disrupted by detergents. Most detergents are amphipathic molecules with a polar head and disordered hydrocarbon tail. Aβ is also amphipathic, its N-terminus region is polar and its C-terminus region is hydrophobic. Aβ40 oligomers may well be relatively disordered in water as they disrupt neuronal membranes in a manner resembling that caused by detergents (15). In contrast, Aβ42 forms hexamers, dodecamers, and octamers in water with a high content of β secondary structure (16) and at least four distinct sizes of stable channels in neurons (15). Aβ42 channels have been reported to have β-barrel structures (17).
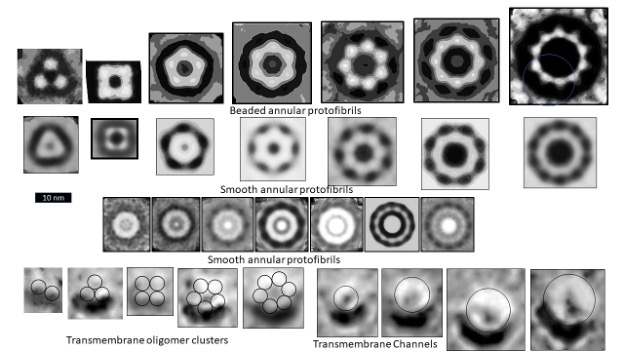
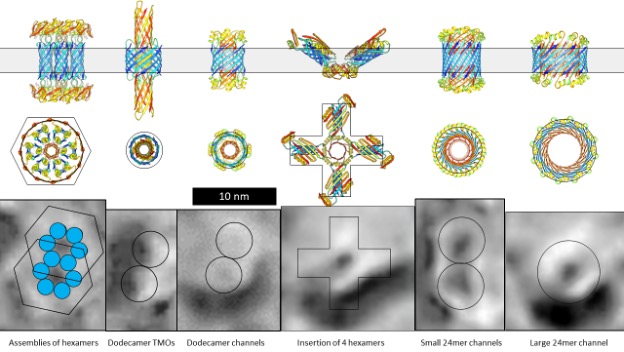
Figure 1 illustrates electron microscopy images of three categories of Aβ42 Annular Protofibrils (APFs) (14) and two categories of transmembrane assemblies (14). The beaded APFs form first when globular Aβ42 assemblies are dried on a microscopic surface in the presence of hexane. These APFs resemble beads of necklaces. At first glance the micrographs appear indecipherable; the number of beads, the sizes of the beads, and the shapes of the APFs vary, and some beads appear to have merged. Upon closer examination, the beads can be classified into six sizes. Based on other experimental findings, we postulate that these beads correspond to oligomers with 6, 8, 12, 16, or 18 peptides. In some images, only one size of bead comprises relatively symmetric APFs. We first radially averaged these images and then averaged multiple images of the same size and shape. The first row of Figure 1 shows averaged images of APFs formed by the smallest and most abundant size of beads, hexamers in our models.
But beaded APFs are not static; they gradually morph into smooth APFs. Two categories of smooth APFs are illustrated, one with a black center and one with a white center. Those with black centers are more like the beaded-APFs from which they develop. In our concentric β-barrel models explained below, they have the same number of subunits as the beaded APFs above them. Concentric rings are more apparent in the white-centered APFs of the third row. These images are the best visual evidence for concentric β-barrel structures. Each ring corresponds to one of three concentric β-barrels in our models, a middle hydrophobic barrel is sandwiched between two barrels with water-exposed hydrophilic surfaces. The number of subunits comprising our models of the white-centered APFs increase in steps of four from 24 to 48 subunits.
When a lipid bilayer is frozen the two lipid leaflets can be split apart to reveal the presence and shape of proteins that span the membrane. The first five freeze-fracture images of transmembrane Aβ42 assemblies (13) resemble beaded APFs with two to six large beads. Similar images of portions of Aβ assemblies that extend above the bilayer have been observed by atomic force microscopy (18). We suspect that each peak corresponds to a transmembrane oligomer (TMO) composed of 12 or 16 peptides. The next four images, from the same micrograph, resemble the smooth APFs. We propose that these are ion channels that have morphed from the TMO aggregates. Freeze-fracture images of more transmembrane assemblies will be presented later.
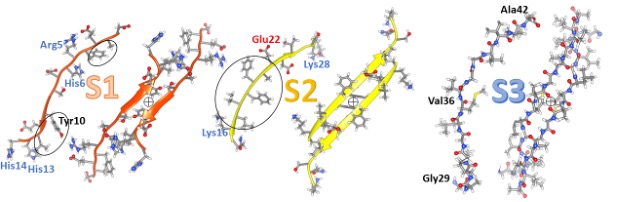
Our model of a soluble hexamer in Figure 2 resembles one we proposed previously (12). We classify the Aβ42 sequence into three segments (S1, S2, and S3) of about the same length, each of which forms a β-strand in these models. β-strands are the most elongated form of a polypeptide chain; side chains of every other residue are on opposite sides of the strands as can be seen in side-views. Amino acid side-chains can be classified into three categories according to their distribution between the surface and interior of soluble proteins or their partition energies between water and organic solvents: hydrophilic (water-loving) side-chains favor a polar aqueous environment, hydrophobic (water-avoiding) side-chains prefer an apolar environment, and ambivalent side-chain don’t much care (19). S1 is the hydrophilic of the segments; eight of its fourteen residues have charged side chains most of which are on odd-numbered residue positions. Its four hydrophobic residues are circled in the figure and occur at even-numbered positions between charged residues. S1 is disordered in some fibril structures but forms one or two β-strands in others. It is the most difficult segment to model but may be the most important for drug development. Mutations in rat Aβ42 at residues arginine 5, tyrosine 10, and histidine 13 eliminate its toxicity and ability to form channels. Two findings suggest that two S2 segments interact to form an antiparallel pair: (1) under oxidizing conditions, centrally located tyrosine 10 side-chains cross-link to form dimers (20), and (2) copper ions bind between side-chains of histidine 6 and histidine 13 and/or histidine 14 (21). The circled plus symbol between the orange S1 strands indicates a P2 rotational axis of 2-fold symmetry. In some other models, S1 forms a double-stranded S1a-S1b β-hairpin with the connecting turn occurring in the polar central region. S1a is the least conserved region, followed by S1b.
S2 segments comprise parallel β-sheets in most fibrils; however, they comprise an antiparallel sheet in fibrils of the pathogenic Iowa mutant (22). S2 has five consecutive hydrophobic residues, circled in the figure, flanked by a positively charged lysine 16 and a negatively charged glutamate 22. In fibrils and most of our models, three of the hydrophobic side chains are on the buried side of the sheet, and the charged side chains are on the water-exposed side. Most S3 residue positions are identical among terrestrial vertebrate sequences. In some other models, S2 forms an amphipathic α-helix with hydrophobic side chains on one side and primarily charged side chains on the other side. This ability to adopt either helical or beta secondary structure is also a property of the N-terminus region of synucleins.
S3 is the most hydrophobic segment, no polar side-chain atoms occur from glycine 28 through alanine 42. It is also the most highly conserved; mutations between bony fish and other vertebrates occur only in three positions and these mutations are conservative replacements among hydrophobic alkyl side chains. All backbone atoms of S3, including the polar oxygen and nitrogen of the amide linkages, are shown in Figure 2. β-strands interact to form β-sheets due to the formation of hydrogen bonds between backbone oxygens with partial negative charges (red spheres) and hydrogen-attached nitrogens (blue spheres) with partial positive charges. These hydrogen bonds are vital for membrane-spanning β-barrels since there are few other hydrogen bonding possibilities due to the absence of water and the paucity of polar side chains. β sheets have a cloth-like structure; in fact, silk is composed of β-sheets. For positions of proteins embedded in the alkyl phase of membranes, energetically unfavorable lipid exposure of polar backbone atoms at the edges of the sheets can be avoided by rolling the sheets into cylinders; i.e., β-barrels.
The lower portion of the figure illustrates two views of our hexamer model. This model has 3-fold radial symmetry about the central axes of the barrels and 2-fold perpendicular P2 symmetry. This symmetry requires all six subunits to have the same structure and interaction. It, along with well-established β-barrel theory, places severe constraints on β-barrel models, especially if each subunit contributes only one or two strands per barrel. Concentric β-barrel structures are even more constrained because symmetry axes must correspond for all barrels, and distances between the walls of adjacent barrels can be limited between 0.7 and 1.2 nm.
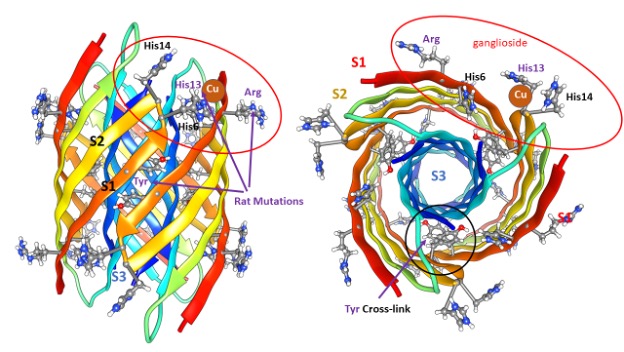
S3 segments comprise a hydrophobic 6-stranded antiparallel β-barrel buried in the core. S1 and S2 strands comprise a surrounding 12-stranded antiparallel β-barrel. Electrostatic interactions are near-optimal: almost all charged side-chains are exposed to water on the surface and all 42 negatively charged carboxyl groups of aspartate, glutamate, and the C-termini form salt bridges with 42 positively charged groups of lysine, arginine, and histidine side-chains and the N-terminus amine. All other polar atoms form hydrogen bonds with one another or with water on the surface.
Except for two S2 side chains per subunit, all hydrophilic side chains are at least partially buried. Some features of this structural motif were unprecedented when first proposed but have since been observed experimentally; (1) Antiparallel six-stranded hydrophobic β-barrel structures called cylindrin with 3fold and 2-fold symmetries have been solved (23), (2) NMR studies of oligomers with four subunits (24) and 32 subunits (25) have demonstrated that S3 segments form antiparallel β-pairs with P2 symmetry between adjacent valine 36 residues as occurs in our models, (3) concentric β-barrel structures have been solved in toxin proteins (26, 27,) that are composed of seven or nine identical subunits, and (4) NMR studies have shown that Aβ42 channels have β-barrel structures (17). Although not a feature of our original models, recent studies indicate that in biological membranes formation of Aβ42 channels is facilitated by a ganglioside lipid (Monosialotetrahexosylganglioside – that Mary Poppins would be unable to pronounce) commonly called GM1. The extremely large head group of GM1 with its five sugar-like rings has been reported to interact with side-chains of arginine 5 (5), histidine 13, and histidine 14 (6). The red ovals indicate that these residues are proximal in our hexamer models and this interaction has been included in some of our Aβ42 channel models.
Figure 3 illustrates six of our models of how Aβ42 hexamers interact with and span lipid membranes to form TMOs and interact to form channels composed of a multiple of six peptides. Side views are in the first row, views down the central axis are in the second, and freeze-fracture images with the approximate shape and dimensions of the models are in the third. In the first model, only S3 β-barrels are in the lipid alkyl phase, the S2 secondary structure has become helical and S1 segments form β-hairpins around the coiled-coil bundle of three S2 helices. These hexamers may interact to form hexagonal lattice-type assemblies in which some S1 strands line a pore through the center of a hexamer-of-hexamers. The next two models are dodecamers. S3 segments comprise the outer 12-stranded lipid-exposed barrel. In the first dodecamer half of the S2 segments form an inner 6-stranded barrel that is too small for ions to pass. The other half of the S2 segments interact with S1 β-strands to form a 9-stranded β-barrel that extends well beyond the membrane surfaces. This model is consistent with images of both atomic-force microscopy and freeze-fracture images that have two to six peaks. The next dodecamer model has only one subunit conformation: six S2 helices in each aqueous phase surround a parallel 6-stranded S1 β-barrel. S1a segments extend into the S3 barrel from each side to form a small ion-conducting pore lined with charged side chains. This model is consistent with findings that in some Aβ42 membrane assemblies S2 segments are susceptible to digestion by trypsin in the aqueous phase, but S1 and S3 segments are not (5). Two such dodecamers may interact to form freeze-fracture images with two barely detective pores.
The fourth model depicts our hypothesis of how four hexamers bind to the membrane surface, aggregate, and then penetrate the bilayer. Each hexamer’s structure is like that of the soluble hexamer except that two-thirds of the S1-S2 segments have peeled away from the S3 barrel, allowing much of its hydrophobic surface to interact with the top lipid leaflet. Next S1-S2 segments of the ends of four hexamers may interact to penetrate the inner leaflet as a frequently observed protein structural motif, a bundle of eight α-helices surrounding an 8-stranded β-barrel. Freeze-fracture images of some channels appear to have 4-fold radial symmetry and dimensions consistent with this model. The final two models each have 24 subunits with identical conformations. As in the dodecamer channel model, all S2 segments form α-helices on each membrane surface that surround a S1 β-barrel. In the smaller diameter 24mer channel, S1s are single strands that form a 12-stranded parallel β-barrel; the S1a portion of these two barrels fit inside the outer 24-stranded S3 β-barrel to line the pore. The S3 strands of the larger diameter barrel are more tilted, the S2 helices are farther into the membrane surface, and S1a-S1b β-hairpins comprise 24-stranded antiparallel β-barres that line the pore. Increased tension on the membrane could cause a transition from the smaller to the larger 24mer channel. If so, these channels would be mechanosensitive, like other channels of known structure that we have modelled (28,29).
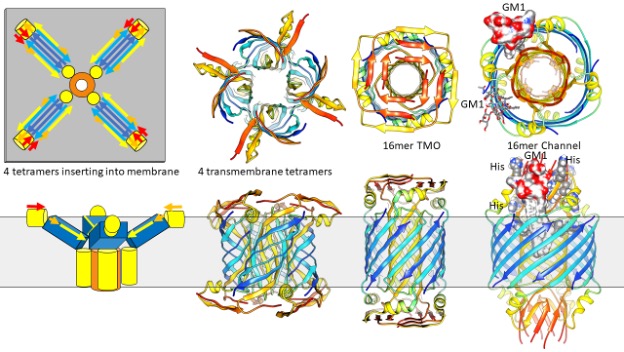
Not to complicate matters, but other equally viable models are illustrated in Figure 4. NMR experiments. indicate that in detergents, Aβ42 tetramers comprise a 6-stranded antiparallel β-sheet (24). These tetramers have two subunit conformations. S3 segments comprise the two central β-strands. An axis of P2 rotational symmetry is located between valine 36 residues of this pair, as in our hexamer models. S1 and S2 segments of these subunits were disordered. S2-S3 segments of the other subunits form a β-hairpin with S3 binding to the central S3 strands and S2 on each edge. Figure 4 illustrates our models of how these types of tetramers could bind to and then penetrate membranes to form concentric β-barrel assemblies that have a multiple of four peptides. The shapes and sizes of these models are similar to those of Figure 3, so it is difficult to predict which type of model is better based on the EM images. A major difference between these models and most of those in Figure 3 is that half of the S2 strands form the inner pore-lining β-barrel while the other half form α-helices on the membrane surfaces. S1 segments form β-structures that may be atop the S2 helices as in the 16mer TMO model or be surrounded by S2 helices as in the 16mer channel model. As in the 24mer models, S3 strands of the larger diameter assembly are more tilted than those of the smaller diameter assembly, an increase in membrane tension could cause a transition from the closed TMO conformation to an open channel.
The most interesting aspect of the 16mer channel model is that it provides a mechanism by which GM1 lipids could bind and keep the channel in an open conformation. GM1 lipids have been docked into these gaps so that their negatively charged carboxyl group binds to positively charged arginine 5, histidine 13, histidine 14 side chains proposed to be involved in GM1 binding. To our surprise, there is sufficient space between the outer S3 barrel and pore-lining S2 barrel to accommodate the two alkyl chains of GM1.
We are currently developing models of interactions among different amyloids; i.e. of both toxic and protective assemblies formed when Aβ42 interacts α-synuclein, amylin, a peptide cleaved from PRP prions, humanin, and cystatin C. Understanding such structures could be useful in understanding associated diseases and ways to treat or prevent them. Structures of these more complex assemblies may be easier to solve because they are likely to be less polymorphic.
Structure-based drug design requires knowing which structures to target, when to target them, and the determination of their structures. If we want to nip Alzheimer’s disease in the bud, we should inhibit the formation of toxic assemblies at or before the mild cognitive stage. It may be too late if we wait until plaques and tangles form. We still have much to do to understand the structures of both functional and toxic amyloid assemblies. Our models are detailed hypotheses intended to be tested rather than believed. Possible ways to do that are too numerous to list here. Our models also point out what additional experiments should be funded and performed. The micrographs shown here were developed over a decade ago. The freeze-fracture micrographs were not published at first and the study was terminated because the micrographs were considered too complicated to interpret. I suspect that has occurred in other labs as well. We now understand that amyloid-associated diseased likely involve interactions of oligomers with membranes and that at least some membrane assemblies are probably intrinsically diverse rather than intrinsically disordered. If so it should be possible to obtain much-needed higher-resolution results using contemporary methods such as cryo-microscopy and single particle analyses.
The goal of understanding the precise structures of these assemblies is not hopeless. It’s well past time to support these efforts. High-resolution structures could prove invaluable for the development of effective preventions, treatments, and cures. Just imagine how much that would reduce human suffering and medical costs!
Computer graphics of molecular models were generated using UCSF Chimera. http://www.rbvi.ucsf.edu/chimera
References
- Cline EN, Bicca MA, Viola KL, Klein WL. The Amyloid-beta Oligomer Hypothesis: Beginning of the Third Decade. J Alzheimers Dis. 2018;64(s1):S567-S610.
- Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012 Jul;2(7):a006338. doi: 10.1101/cshperspect.a006338. PMID: 22762015; PMCID: PMC3385944
- Patricia Sinclair 1, Ancha Baranova 2 and Nadine Kabbani 2,* Mitochondrial Disruption by Amyloid Beta 42 Identified byProteomics and Pathway Mapping International Journal o fMolecular Sciences Cells 2021, 10, 2380. https://doi.org/10.3390/cells10092380 https://www.mdpi.com/journal/cells
- Durell SR, Guy HR, Arispe N, Rojas E, Pollard HB. Theoretical models of the ion channel structure of amyloid beta-protein. Biophys J. 1994 Dec;67(6):2137-45. doi: 10.1016/S0006-3495(94)80717-9. PMID: 7535109; PMCID: PMC1225600.
- Dong Yan Zhang, Jian Wang, Rebecca M. Fleeman, Madison K. Kuhn, Matthew T. Swulius,Elizabeth A. Proctor, and Nikolay V. Dokholyan*. Monosialotetrahexosylganglioside Promotes Early Aβ42 Oligomer Formation and Maintenance ACS Chem. Neurosci. 2022, 13, 1979-1991
- Jacques Fantini | Henri Chahinian | Nouara Yahi. Progress toward Alzheimer’s disease treatment: Leveraging the Achilles’ heel of Aβ oligomers? The Protein Society, Protein Science. 2020;29:1748–1759.
- Elena Hernando-Pérez 1,2 , Victor Tapias 1,2 , María Calvo-Rodríguez 1,2,†, Carlos Villalobos 1,* and Lucía Núñez 1,2,*. Amyloid Beta Oligomers-Induced Ca2+ Entry Pathways: Role of Neuronal Networks, NMDA Receptors and Amyloid Channel Formation Erica Caballero 1,2, Biomedicines 2022, 10, 1153.
- Meade RM, Fairlie DP, Mason JM. Alpha-synuclein structure and Parkinson’s disease – lessons and emerging principles. Mol Neurodegener. 2019 Jul 22;14(1):29.
- Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, Moir RD. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016 May 25;8(340):340ra72. doi: 10.1126/scitranslmed.aaf1059. PMID: 27225182; PMCID: PMC5505565.
- Shafrir, Y., Durell, S., Arispe, N. & Guy, H. R. Models of membrane-bound Alzheimer’s Abeta peptide assemblies. Proteins 78, 3473-3487, doi:10.1002/prot.22853 (2010).
- Shafrir, Y., Durell, S. R., Anishkin, A. & Guy, H. R. Beta-barrel models of soluble amyloid beta oligomers and annular protofibrils. Proteins 78, 3458-3472, doi:10.1002/prot.22832 (2010).
- Yun, S. J., Yun, S. J. & Guy, H. R. Analysis of the stabilities of hexameric amyloid-beta(1-42) models using discrete molecular dynamics simulations. J Mol Graph Model 29, 657-662, doi:10.1016/j.jmgm.2010.11.008 (2011).
- Durell SR, Guy HR. The amyloid concentric β-barrel hypothesis: models of Synuclein oligomers, annular protofibrils, lipoproteins, and transmembrane channels. Proteins: Structure, Function, and Bioinformatics. 2021:1-31.
- Durell SR, Kayed R, Guy HR. The amyloid concentric β-barrel hypothesis: Models of amyloid beta 42 oligomers and annular protofibrils. Proteins. 2022 May;90(5):1190-1209. doi: 10.1002/prot.26301. Epub 2022 Jan 25. PMID: 35038191; PMCID: PMC9390004
- Bode, D. C., Baker, M. D. & Viles, J. H. Ion Channel Formation by Amyloid-β42 Oligomers but Not Amyloid-β40 in Cellular Membranes. Journal of Biological Chemistry 292, 1404-1413, doi:10.1074/jbc.M116.762526 (2017).
- Yoshiike, Y., Kayed, R., Milton, S. C., Takashima, A. & Glabe, C. G. Pore- forming proteins share structural and functional homology with amyloid oligomers. Neuromolecular medicine 9, 270-275 (2007).
- Serra-Batiste, M. et al. Aβ42 assembles into specific β-barrel pore- forming oligomers in membrane-mimicking environments. Proceedings of the National Academy of Sciences 113, 10866-10871, doi:10.1073/pnas.1605104113 (2016).
- Quist, A. et al. Amyloid ion channels: a common structural link for protein- misfolding disease. Proc Natl Acad Sci U S A 102, 10427-10432, doi:10.1073/pnas.0502066102 (2005).
- Guy HR. Amino acid side-chain partition energies and distribution of residues in soluble proteins. Biophys J. 1985 Jan;47(1):61-70. doi: 10.1016/S0006-3495(85)83877-7. PMID: 3978191; PMCID: PMC1435068.
- Urbanc B. Cross-Linked Amyloid beta-Protein Oligomers: A Missing Link in Alzheimer’s Disease Pathology? J Phys Chem B. 2021;125(5):1307- 1316.
- Williams TL, Serpell LC, Urbanc B. Stabilization of native amyloid β- protein oligomers by Copper and Hydrogen peroxide Induced Cross-linking of Unmodified Proteins (CHICUP). Biochim Biophys Acta. 2016 Mar;1864(3):249-259. doi: 10.1016/j.bbapap.2015.12.001. Epub 2015 Dec 15. PMID: 26699836.
- Qiang, W., Yau, W. M., Luo, Y., Mattson, M. P. & Tycko, R. Antiparallel beta-sheet architecture in Iowa-mutant beta-amyloid fibrils. Proc Natl Acad Sci U S A 109, 4443-4448, doi:10.1073/pnas.1111305109 (2012).
- Do, T. D. et al. Amyloid β-Protein C-Terminal Fragments: Formation of Cylindrins and β-Barrels. Journal of the American Chemical Society 138, 549-557, doi:10.1021/jacs.5b09536 (2016).
- Ciudad S, Puig E, Botzanowski T, et al. Abeta(1-42) tetramer and octamer structures reveal edge conductivity pores as a mechanism for membrane damage. Nat Commun. 2020;11(1):3014.
- Gao Y, Guo C, Watzlawik JO, et al. Out-of-Register Parallel beta-Sheets and Antiparallel beta-Sheets Coexist in 150-kDa Oligomers Formed by Amyloid-beta(1-42). J Mol Biol. 2020;432(16):4388-4407.
- Iacovache, I. et al. Cryo-EM structure of aerolysin variants reveals a novel protein fold and the pore-formation process. Nat Commun 7, 12062, doi:10.1038/ncomms12062 (2016).
- Bokori-Brown, M. et al. Cryo-EM structure of lysenin pore elucidates membrane insertion by an aerolysin family protein. Nat Commun 7, 11293, doi:10.1038/ncomms11293 (2016).
- Sukharev S, Betanzos M, Chiang CS, Guy HR. The gating mechanism of the large mechanosensitive channel MscL. Nature. 2001 Feb 8;409(6821):720-4. doi: 10.1038/35055559. PMID: 11217861.
- Milac A, Anishkin A, Fatakia SN, Chow CC, Sukharev S, Guy HR. Structural models of TREK channels and their gating mechanism. Channels (Austin). 2011 Jan-Feb;5(1):23-33. doi: 10.4161/chan.5.1.13905. Epub 2011 Jan 1. PMID: 21084863; PMCID: PMC3052205.
Read and download this full eBook here: ‘Why are the root causes of amyloid-associated diseases so misunderstood and treatments so inadequate?’